Python In class challenge answers 10-26-18
Here are the answers to the python challenges this week in class. The formatting isn’t great here, so the link below will show another format if it’s which might be easier for you to understand/read.
https://hackmd.io/s/S1oyAag3Q#
# cumulative sum
x=[3,10,4,12,55]
cs=[0]*5
for i in range(0,len(x)):
cs[i]=sum(x[0:(i+1)])
print(cs)
x=[3,10,4,12,55]
cs=list()
for i in range(0,len(x)):
cs.append(sum(x[0:(i+1)]))
print(cs)
#how long to 5 5’s
x=[5,3,2,5,5,1,2,5,3,5,1,5,1]
count=0
i=0
while count < 5:
if x[i]==5:
count+=1
i+=1
print(i)
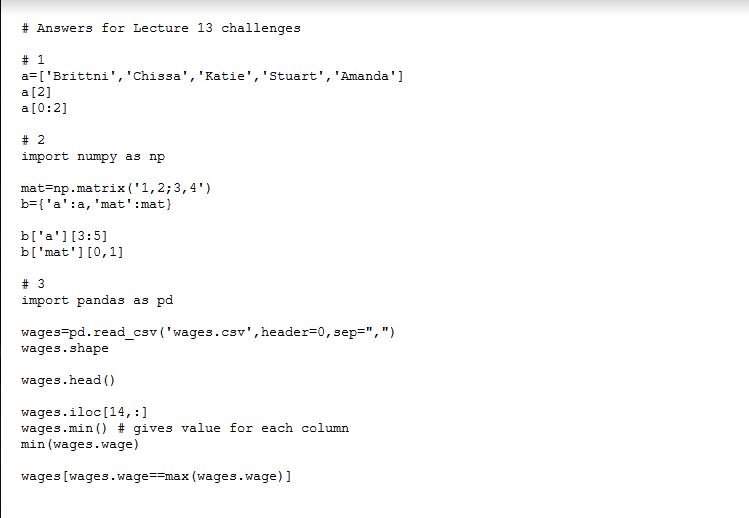
Answers for Lecture 11 extra practice
#
import pandas as pd
# 0 – calculate sum of female and male wages in wages.csv
wages=pandas.read_csv(“wages.csv”,header=0,sep=”,”)
femaleSum=0
maleSum=0
for i in range(0,len(wages),1):
if wages.gender[i]==”female”:
femaleSum=femaleSum+wages.wage[i]
else:
maleSum=maleSum+wages.wage[i]
femaleSum
maleSum
#the two commands below won’t work – why?
sum(wages.gender==”female”)
sum(wages.gender==”male”)
Find runs –
This is the super abstract one we went over in Tutorial!
# load file – available on Sakai
findRuns=pd.read_csv(“findRuns.txt”,header=None,sep=”\t”)
# create a variable out that is currently undefined
out=pd.DataFrame(columns=[‘startIndex’,’runLength’])
# I will use this variable cur to hold onto the previous number in the vector;
# this is analagous to using findRuns[i-1]
cur=findRuns.iloc[0,0]
#cur=findRuns[i-1]
# this is a counter that I use to keep track of how long a run of repeated values is;
# if there are not repeated values than this count equals 1
count=1
# loop through each entry of our vector (except the 1st one, which we set to cur above)
for i in range(1,50,1):
# test if the ith value in the vector findRuns equals the previous (stored in cur)
if findRuns.iloc[i,0]==cur:
# test whether count is 1 (we aren’t in the middle of a run) or >1 (in the middle of a run)
if count==1:
# if the ith value in the vector equals the previous (stored in cur) and count is 1, we
# are at the beginning of a run and we want to store this value (we temporarily store it in ‘start’)
start=(i-1)
# we add one to count because the run continued based on the ith value of findRuns being equal to
# the previous (stored in cur)
count=count+1
# if the ith value in findRuns is not the same as the previous (stored in cur) we either are not in a run
# or we are ending a run
else:
# if count is greater than 1 it means we were in a run and must be exiting one
if count>1:
# add a row to ‘out’ that will hold the starting positions in the first column and the length
# of runs in the second column; this appends rows to out after finding and counting each run
out.loc[len(out)]=[start,count]
# reset count to 1 because we just exited a run
count=1
# remember cur holds the previous element in findRuns, so we need to update this after each time
# we go through the for loop
cur=findRuns.iloc[i,0]
cur
out