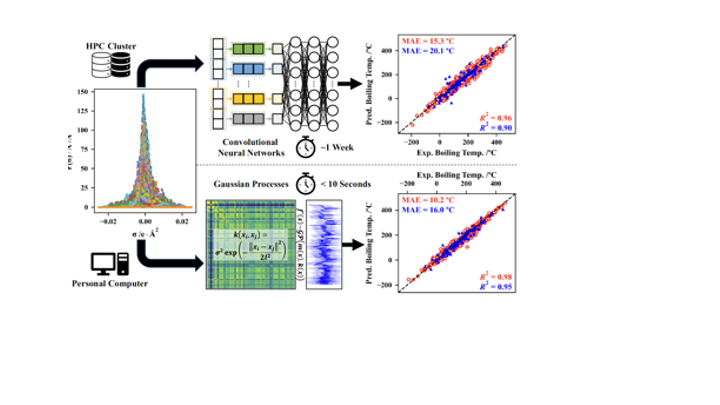
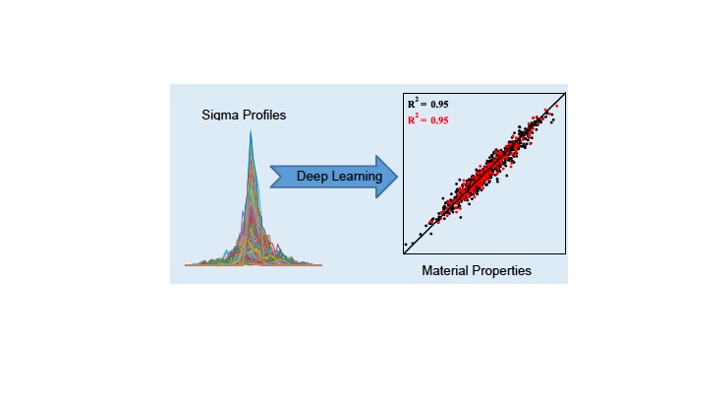
This paper, in collaboration with the Colón group, demonstrates the capability of Gaussian processes to correlate and predict physicochemical properties from sigma profiles. The new approach outperforms state-of-the-art neural networks from earlier studies and, most importantly, can be navigated using standard algorithms to find compounds having desired properties. Check it out here! https://doi.org/10.1073/pnas.2404676121