Compared with natural zeolites, synthetic zeolites comprise over 70% of the global market, which was valued at 29.08 billion USD in 2016. Synthetic zeolites are typically made under hydrothermal conditions using special organic structure-directing agents (OSDAs) that help steer the self-assembly process toward a particular pore shape and Al distribution.
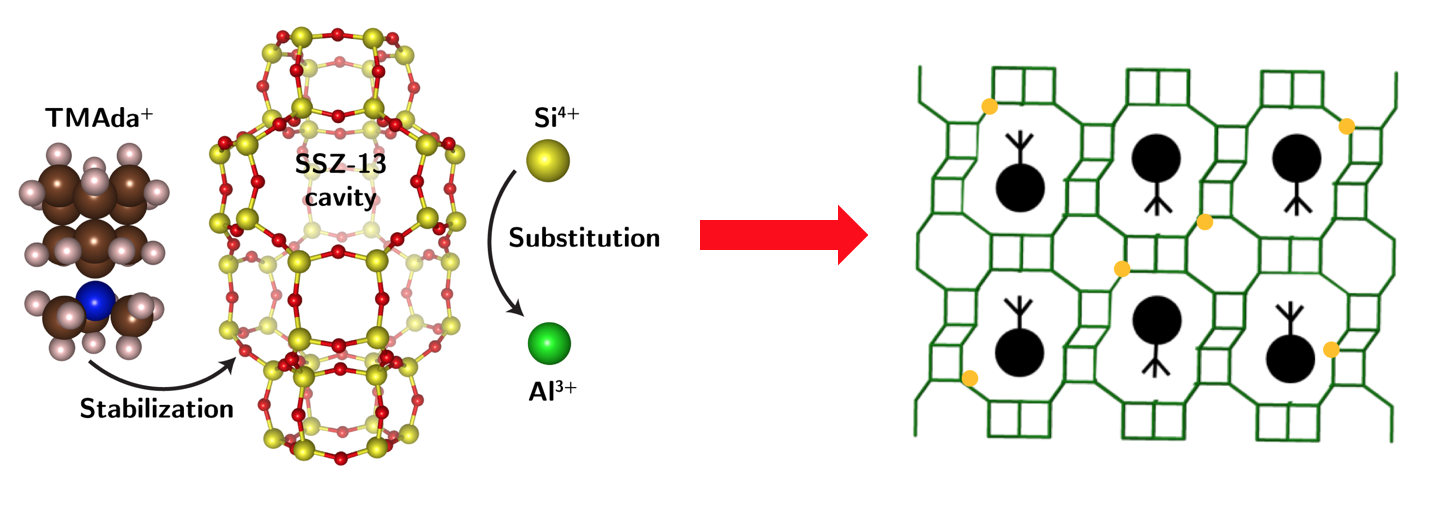
We choose CHA zeolite as a model system, as it presents only a single, symmetry-distinct tetrahedral site, and N,N,N-trimethyl-1-adamantyl ammonium (TMAda+) as an OSDA, as it can adopt only two geometrically equivalent but orientationally opposite configurations within the Chabazite cage.

For the chabazite zeolite framework, two Al atoms can possibly locate in the above distinct arrangements.
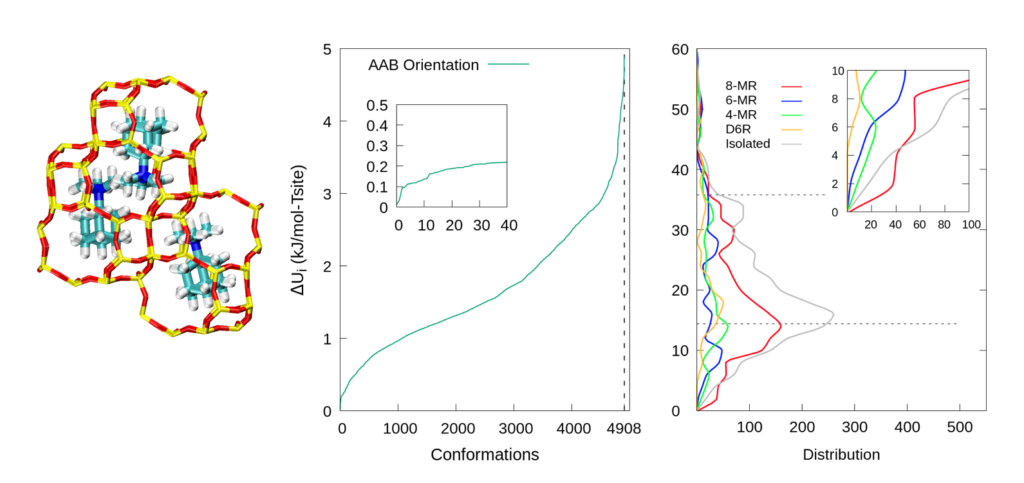
We combined classical and first-principles models to explore the dependence of charged organic structural directing agent (OSDA) with framework (FW) energies on the distribution of Al within the framework. We showed that lattice energies were a strong function of Al distribution for a given orientation of TMAda+. We also found that there is a higher probability of finding a pair of Al atoms on the same 8-member-ring (8MR) than on a 4-member-ring (4MR), a 6-member-ring (6MR), or a double-six-unit (D6R). This result is consistent with experimental observations.
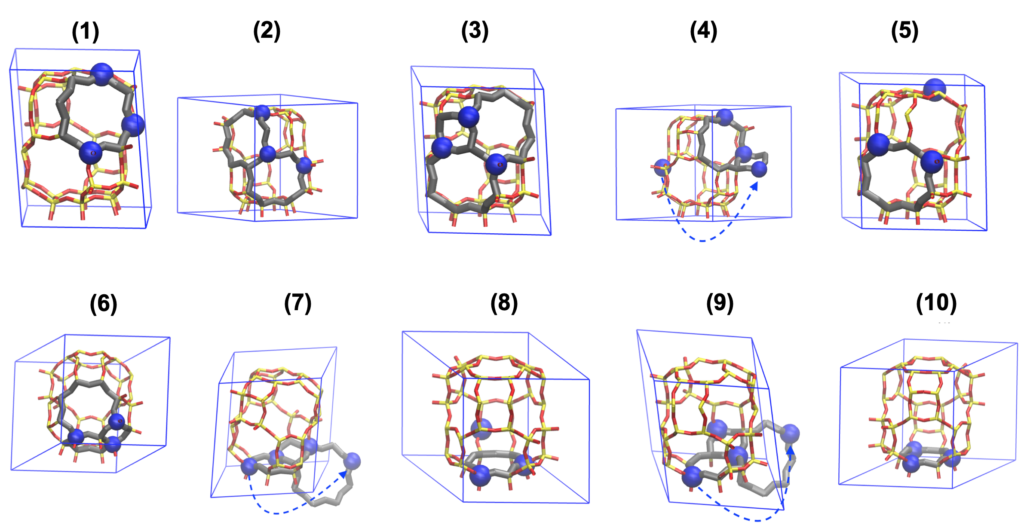
Example framework structures are shown above. Extra frame-work atoms and Al from the nearby image are unwrapped to show a full Al-pair feature. The Al-pair feature is highlighted in gray to show a full ring structure. (1)-(5) are frameworks with lower energies, while (6)-(10) are ones with higher energies.
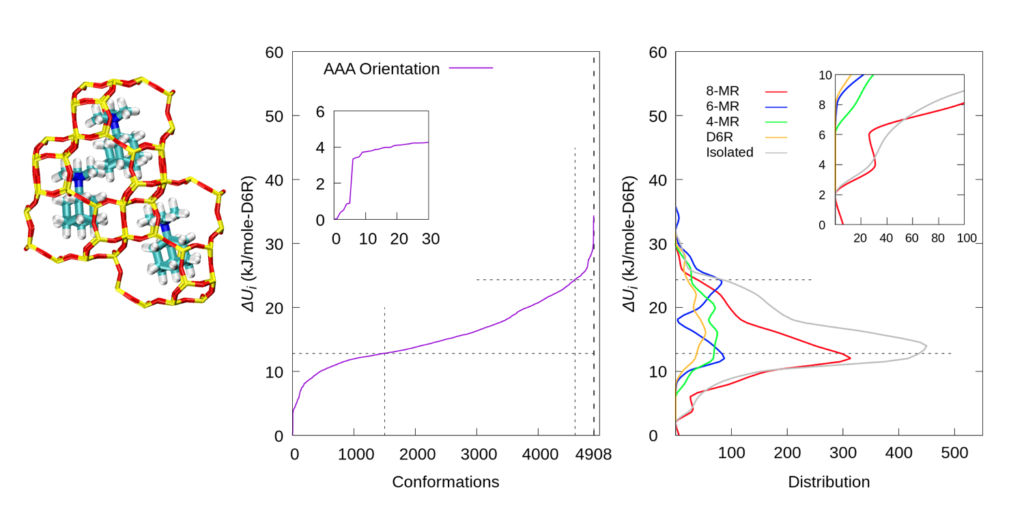
We then changed the orientation of the pre-caged TMAda+s and repeated the procedure to explore all possible Al locations. Similar trends were observed, which indicates that the orientation of pre-caged TMAda+s is not able to change the overall Al distribution.
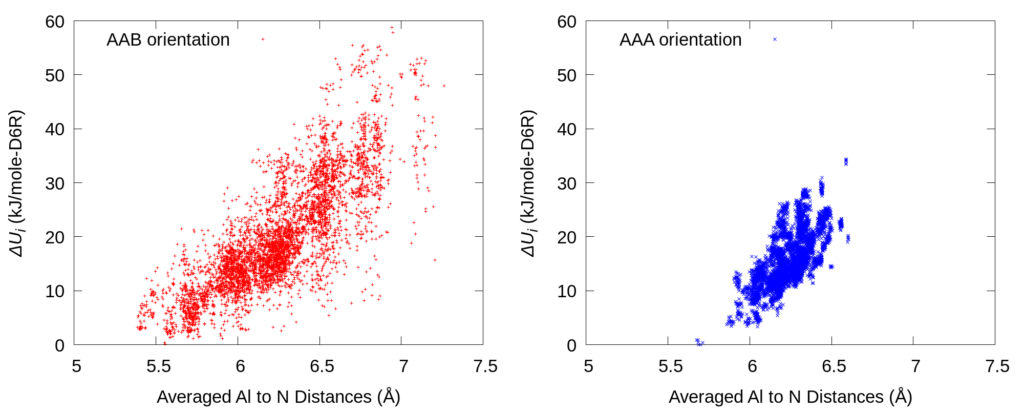
In order to illustrate the underlying relation of Al-to-N (anionic Al site to cationic quaternary ammonium) distance and energy, the above picture shows a parity plot of averaged Al-to-N distances v.s. relative potential energies (∆Ui). The correlation trend indicates that the major contribution to the system energy comes from the cation-to-anion electrostatic interaction.
The highlight of this work is the construction of the classical molecular model, and we have also shown that it can predict some experimental behavior of zeolites, especially the Al distribution. In the future work, we will use the model to: (1) include the inorganic alkali structure-directing agent and investigate its influence on the Al distribution; (2) study other OSDAs and investigate their roles on shaping other aluminosilicate zeolites; (3) develop a coarse grain model to explore more expansively and larger systems.