Introduction and Project Overview
Chemical rocket propellant is a class of materials which provides thrust through the combustion of oxidizer and fuel components. The two most industrially-significant types of rocket propellant are liquid and solid propellants; the oxidizer and fuel chemicals are both in the liquid phase for liquid propellants, while for solid propellants
both components are in the solid phase. Solid phase propellants are utilized due to ease of storage, near instant readiness, and relatively low cost. Liquid phase propellants can have better performance, are throttleable, and have the ability to restart, but can suffer from complications due to handling and storage as well as requiring preparation, such as cryogenic cooling, which introduces significant latency between storage and utilization. For military applications, the advantages of solid propellent often out weigh the better performance of liquid propellant.
Solid energetic compounds used in solid propellants are usually brittle, and thus unable to handle the shock waves present during typical applications. To improve the material properties, the solid energetic compounds, in the form of small crystalline particles, are typically mixed into a viscous pre-polymer which is subsequently cured into a rubbery elastomer possessing the desired material properties. Industrially, a standard solid composite propellant consists of 60–72% ammonium perchlorate (AP) as the oxidizer, up to 22% aluminum powder as metal fuel, and 8–16% polymeric
binder such as hydroxyl-terminated polybutadiene (HTPB)-based polyurethanes, all by percent mass. Due to the unsaturated characteristic of HTPB, over time the cured polymer will undergo thermo-oxidative degradation, continue to cross-link, become brittle, and accordingly lose the desired mechanical properties, leading to potential failure upon use. Even though solid propellants have long shelf lives, their material properties can degrade over the course of a couple decades and the rate of this decay is dependent upon the storage conditions. Once sufficiently aged, there is a non-insignificant risk of device failure upon use due to the formation of defects related to the increase of cross-links in the polymeric binder over time. In particular, microvoids and microcracks associated with embrittlement and debonding of the polymeric binder from the particulate crystalline oxidizer promote uneven burn. Thus, after some empirically determined shelf life devices containing the AP-HTPB propellant are decommissioned.
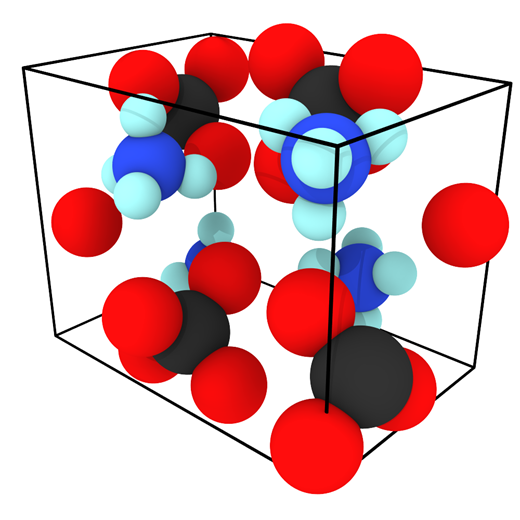
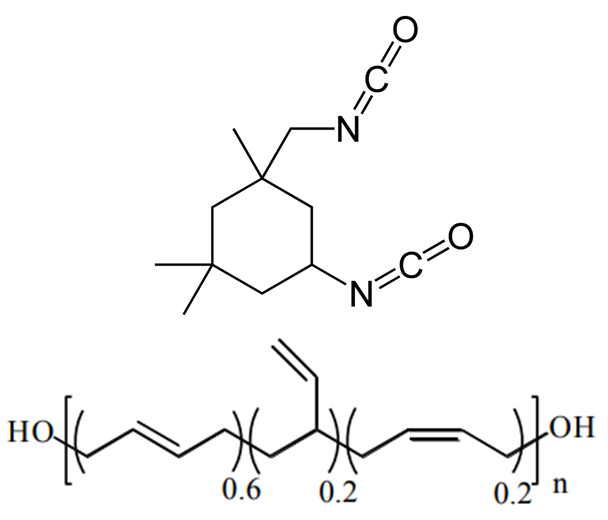
Despite the abundance of knowledge on these materials, the relationship between particular degradation mechanisms and the resulting material properties are not well understood. Experimentally, evaluating an aging process that spans decades in a feasible manner relies upon accelerated aging techniques. These techniques involve exacerbating the conditions favorable to increased cross-linking such as higher-than ambient temperature and higher concentration of oxygen, water, or any other contaminant suspected of inducing cross-linking. The material properties of the propellant can then be sampled over the course of months or a couple years as opposed to decades. Unfortunately, accelerated aging studies inherently do not mimic ambient conditions and thus could favor reaction pathways that would otherwise be relatively uncommon, thereby reducing confidence in the extrapolated aging process. Computer simulations are a potential way to overcome these experimental limitations. Thus far, there has been little to no molecular modeling research on the aging of polymeric binders typically used in solid propellants, and what has been reported is mostly bond dissociation energies used to speculate on decomposition mechanisms. To circumvent temporal constraints, a series of multiscale simulations are envisioned to study the aging reaction mechanisms and rates, the role of interfaces in the aging process, and the mechanical properties of the propellant with different extents of cross-linking.
A quantum–classical framework is intended for these studies to inspect the thermodynamic and kinetic quantities associated with the degradation mechanisms as well as the resulting macroscopically observable material properties obtained as a function of the quantity and type of different degradation products present in the propellant. Quantum mechanical simulations by Mark Gordon’s group at Iowa State University and the Ames Laboratory are being designed to calculate pre-exponential factors and rate constants for a representative set of degradation mechanisms pertaining to both inert and oxidative atmospheres. Edward Maginn’s group at the University of Notre Dame is using classical molecular dynamics to evaluate material properties of degraded propellant via responses to uniaxial tensile and compressive deformation. Eventually, the parameters describing the thermodynamics and kinetics of individual degradation mechanisms will be incorporated into a kinetic Monte Carlo scheme that will be used to provide a model that estimates the composition and material properties of the AP–HTPB propellant as a function of time. It is likely that the atomistic models will need to be mapped onto coarser-grained representations in order to access the long timescales that are characteristic of the multimodal mechanical responses that occur during the deformation and relaxation of elastomeric materials. The first step in realizing this multiscale framework is to identify or develop suitable atomistic models for the species constituting conventional solid rocket propellant. My work to date has been wholly focused on the development of classical models for the AP and HTPB-based polymeric binder.
A Fully-Atomistic Model of HTPB
The OPLS-AA force field was chosen for the fully-atomistic model of HTPB. To validate the OPLS-AA force field for adequately modeling the polymer, comparisons of cis-1,4-polybutadiene (PB) using OPLS-AA to a well-established united-atom model and experimental results were made.
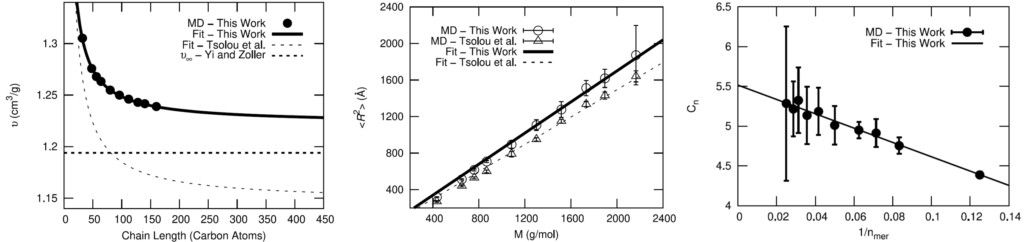
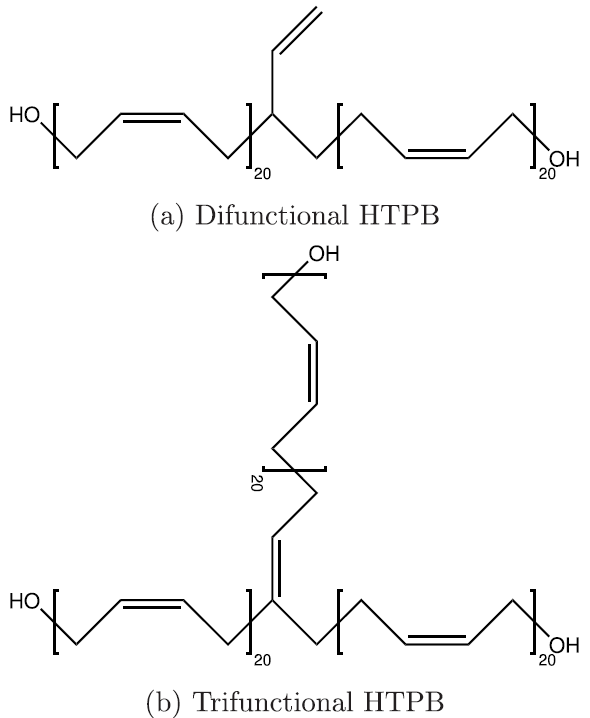
Seeing that the OPLS-AA force field adequately reproduced the volumetric and single-chain structural properties of cis-1,4-polybutadiene, a simple model was developed for HTPB. HTPB is industrially produced by free-radical polymerization which results in a wide distribution of chain sizes and number of hydroxyl functional groups per chain. The average chain weight is 2,800 g/mol and the average number of hydroxyl groups per chain is around 2.5. To approximate this behavior, a mostly cis-1,4 microstructure was used to produce difunctional and trifunctional chain models that when used in a 1:1 ratio succeed in reproducing the average molecular weight and hydroxyl functionality.
The initial configuration of the packed HTPB polymer melt in the simulation box was produced using the proprietary Amorphous Builder Module in the MAPS software package by Scienomics. Subsequently, the polymer was equilibrated using microsecond-long molecular dynamics runs at constant temperature and pressure (T=333 K, P=1 atm). To evaluate the equilibration progress, the running average of the single-chain structural characteristics was monitored over time.
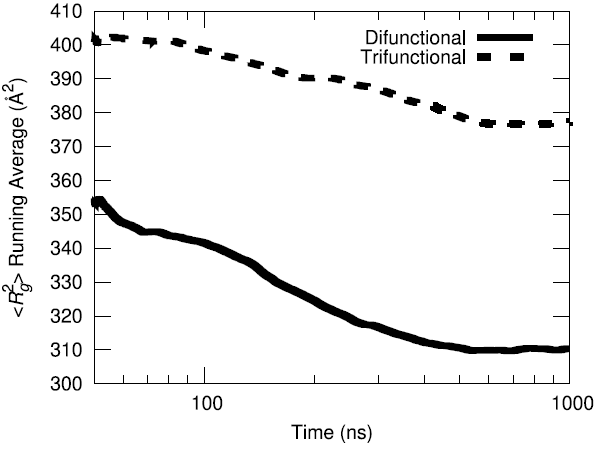
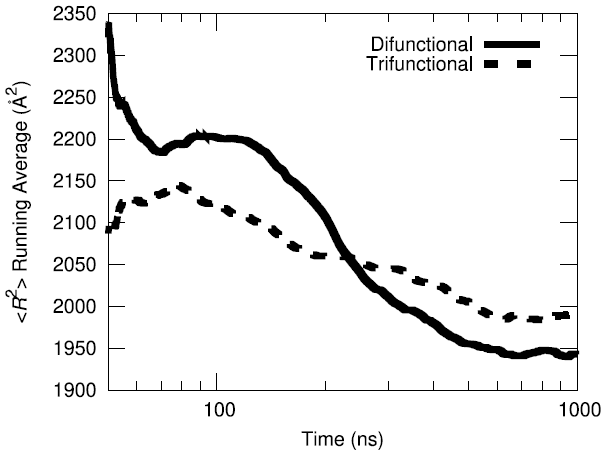
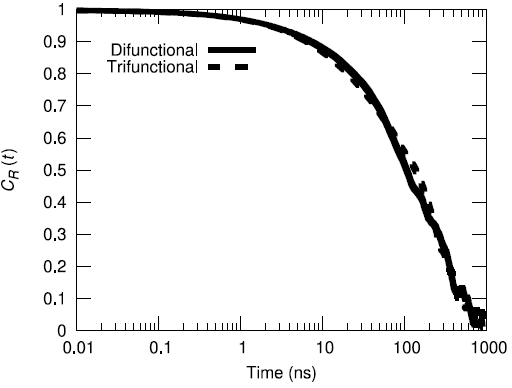
Additionally, the end-to-end vector autocorrelation function was monitored to establish the degree to which the end-to-end vectors decorrelated over the microsecond long trajectory.
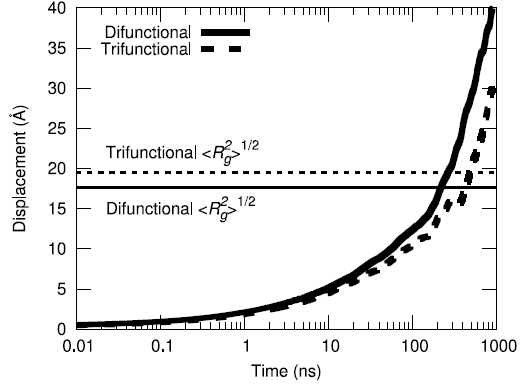
Yet another metric for improving confidence of equilibration is to assess ensemble average of the center-of-mass displacement of the polymer chains to understand how much phase space sampling is occuring. By moving more than their respective radii of gyration on average, both the difunctional and trifunctional chains are shown to have diffused a significant amount during the simulation.
When visualizing the equilibrated simulation cell, significant hydroxyl association was seen to occur. This behavior was unexpected and is not significantly discussed in the existing experimental literature. The infrared spectroscopy data shown in the literature for HTPB suggests that hydroxyl association does occur, but this feature has not been adequately discussed in prior publications.
To quantify the association of the hydroxyl groups, the hydroxyl radial distribution function was computed and the oxygen-oxygen curve indicates that 3.5 Å is a reasonable cutoff distance between oxygen atoms for defining between hydroxyl groups. Using that definition for association, the statistical distribution of hydroxyl associative cluster sizes was calculated.
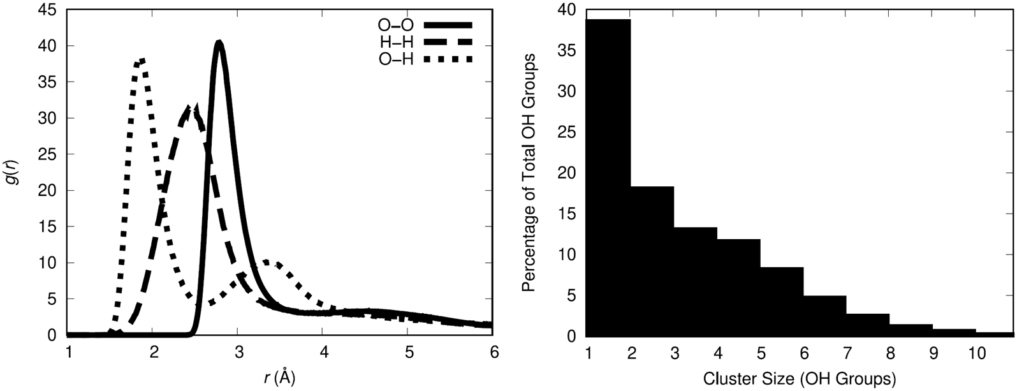
The physical insight gained from these results is that there is a not an even spatial distribution of hydroxyl groups in the polymer melt. This has the potential to influence the distribution of cross-links when the HTPB is cured with the IPDI cross-linking agent. The spatial distribution of cross-links in a cross-linked material impacts the resulting material properties of the cross-linked material. Thus, it is important for a molecular model to adequately represent the spatial distribution of cross-links in the HTPB-IPDI material. Unfortunately, no experimental data exists that characterizes this aspect of the material. Assuming that the hydroxyl groups can serve as an approximate surrogate for the location of cross-links, a cross-linking methodology was developed to maximally retain the OH spatial distribution after cross-linking.
First, hydroxyl pairs had to be identified for cross-linking with an IPDI molecule. To select these pairs, the shortest distance oxygen-oxygen pair was identified, these oxygen atoms removed from the available pool, and then the identification procedure repeated until all 250 hydroxyl groups were paired.
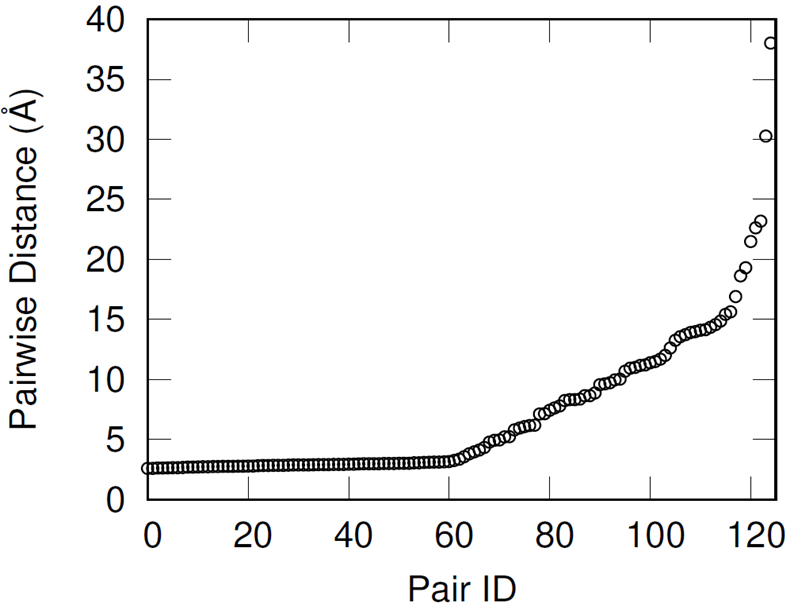
This methodology effectively preserves most of the hydroxyl groups’ associative behavior but has the drawback of producing increasingly distant hydroxyl pairs as the extent of conversion increases (when wanting to cross-link all of the hydroxyl groups). When introducing the IPDI cross-links, the IPDI molecules are directly inserted into the simulation box at the midpoint between reacting hydroxyl pairs and the bonding topology is immediately changed to reflect the cross-linked material. All cross-links are added simultaneously. This procedure results in a significant amount of atomic overlap and very energetic bonds. To relax these unstable configurations in a numerically stable manner, the Fast Inertial Relaxation Engine (FIRE) was utilized to minimize the energy of the system. This minimization algorithm succeeded in reducing atomic overlap and producing nominal-length bonds in the relaxed configurations with just 20,000 iterations.
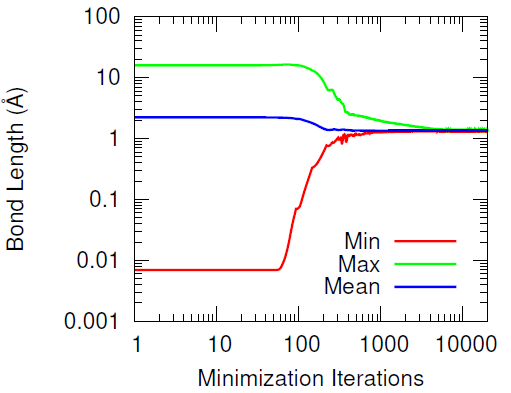
Furthermore, the vast majority of the HTPB atoms were relatively unperturbed during the minimization procedure. This was quantified by computing the displacement of the carbon and oxygen atoms before and after the minimization algorithm. Only a handful of HTPB atoms moved more than 4 Å and the atoms that did were likely connected to the few hydroxyl pairs that were initially separated by large distances.
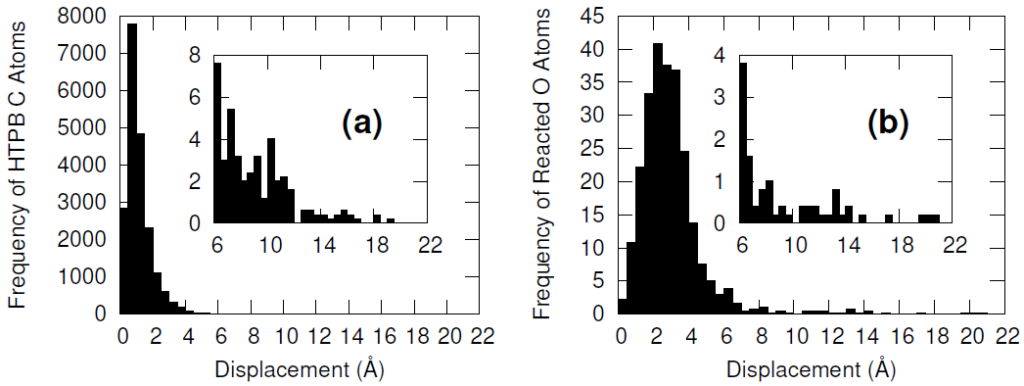
The systems were cross-linked at various isocyanate to hydroxyl ratios (NCO/OH = 0.88, 0.92, 0.96, 1.00) and then subsequently quenched to various temperatures (T in K= 233, 253, 273, 293, 313) and then NPT molecular dynamics was performed for 100 ns to relax stresses remaining after the FIRE minimization algorithm. After the 100 ns of relaxation, the various systems were subjected to uniaxial compressive and tensile deformations to assess the mechanical properties of the HTPB-IPDI cross-linked material. A true strain rate of 107 s-1 was utilized until a 3% strain was achieved at which point the strained dimension was held constant while the lateral dimensions could fluctuate to relax the generated stress. Using this procedure, the temperature dependence of the stress–strain behavior, Young’s modulus, and the stress relaxation behavior was computed.
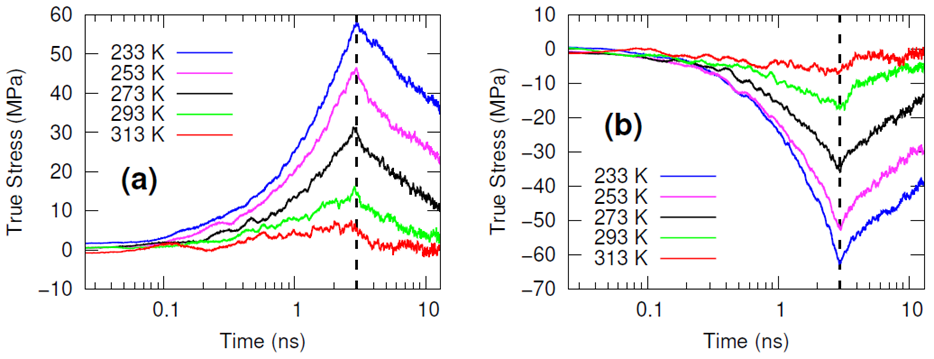
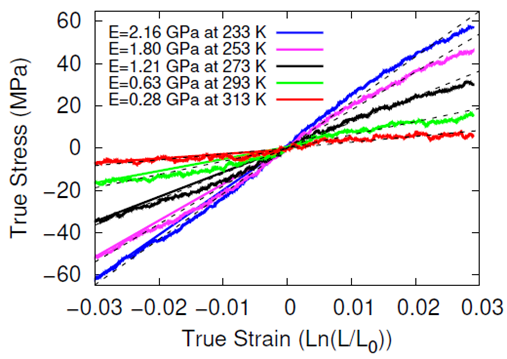
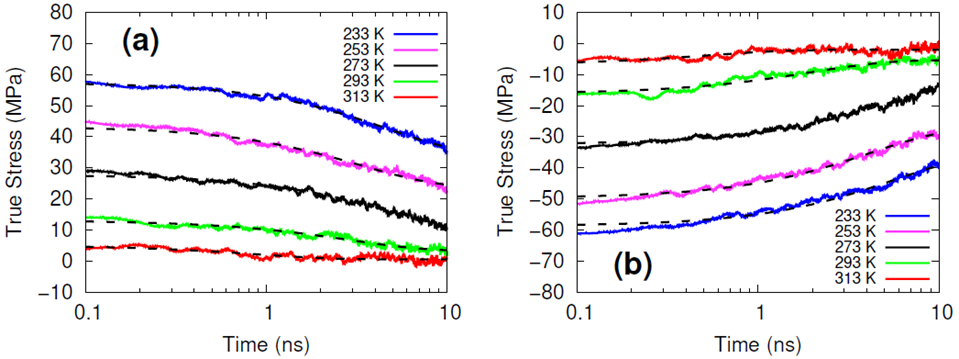
Simulations at the various NCO/OH ratios were indistinguishable within the noise of the results; physical experiments contrast starkly with this observation by showing that there is an extreme sensitivity of the mechanical properties to the NCO/OH ratio. The simulated insensitivity coupled with the simulated true stress relaxation behavior indicates that the current time scale for assessing the mechanical properties via simulation is inadequate for probing the multimodal stress relaxation that occurs for the material. Simply put, the atomistic model for the polymeric binder is too computationally expensive to run simulations long enough to adequately gather statistics on the modes of stress relaxation that occur very slowly. Future work is needed to train heavily coarse-grained model of HTPB that reproduces the atomistic details shown here, specifically the associative behavior of the hydroxyl groups.
Evaluating an Atomistic Model for Ammonium Perchlorate
An existing Class II force field was taken from the literature (Zhu et al.) to describe the bonded and non-bonded interactions of ammonium perchlorate. The existing force field was not well-validated in the original publication and what validation was performed was for the material at 10 K. To gain confidence that the force field performs well for describing the physical properties of AP, several material properties were calculated at 10, 78, and 298 K and compared to experimental data. First, the lattice parameters and density of AP were computed and compared to experiment and the original force field publication as a function of the cutoff radius used for interatomic interactions.
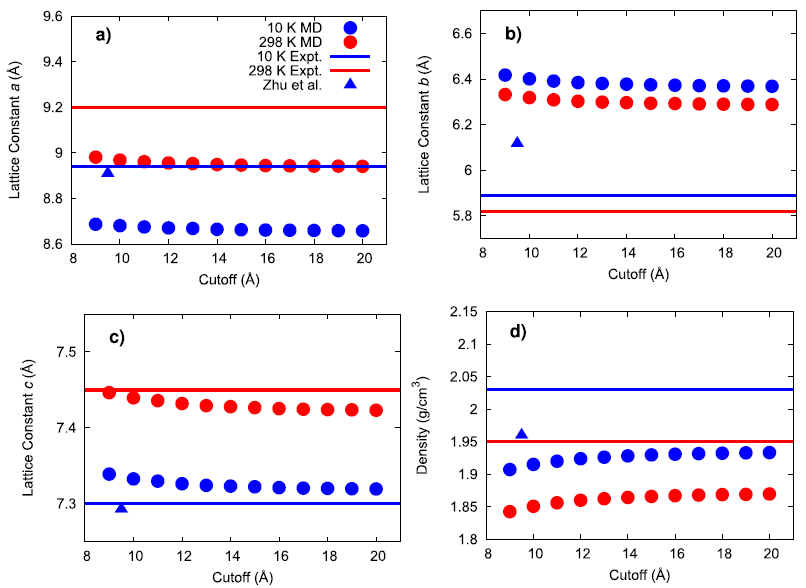
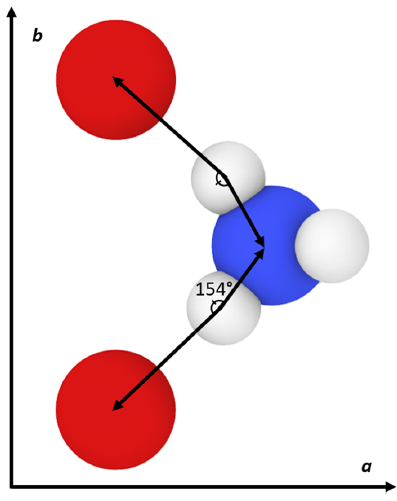
The results generally show that my calculated values do not exactly match the original force field publication’s values, with the exact reason for this discrepancy being unknown. Compared to experiment, my simulated lattice constant a was ~3% too small, lattice constant b was ~8% too large, and lattice constant c was within ~0.4% of the expected value. When inspecting the hydrogen bonding behavior in the system, the simulated N–H(3)•••O(3) mirror symmetric hydrogen bonds were found to have a much larger angle than what was experimentally observed. It is possible that this larger hydrogen bonding angle and the large error in lattice constant b are both manifestations of an underlying inadequacy of the force field since this hydrogen bond type lies in the plane of the hydrogen bonds.
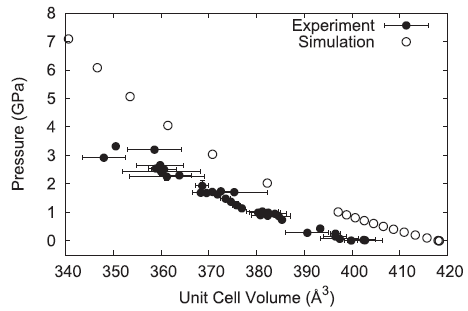
The pressure–volume behavior of the simulated ammonium perchlorate generally reproduces the experimentally observed trend but is overly repulsive resulting in a larger volume for a given pressure.
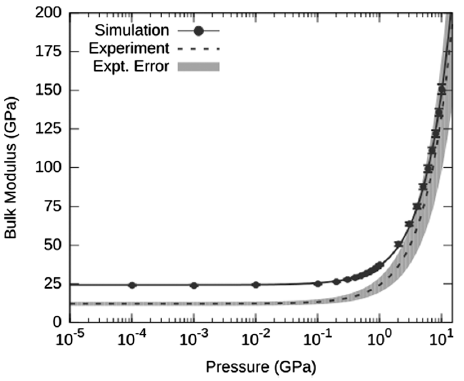
This behavior is similarly seen when viewing the data in terms of the bulk modulus of the AP crystal as a function of pressure.
To evaluate the vibrational behavior of the ammonium perchlorate ions, the 298 K infrared spectrum obtained via experiment was compared to the molecular vibrations obtained from simulation. The stretching and scissoring modes in the simulation were identified for both the perchlorate and the ammonium ions by utilizing bond and angle autocorrelation functions. It was generally observed that the simulated model well reproduces the general locations of the peaks, but the N–H stretching mode was somewhat faster in the simulations than what was observed by experiments.
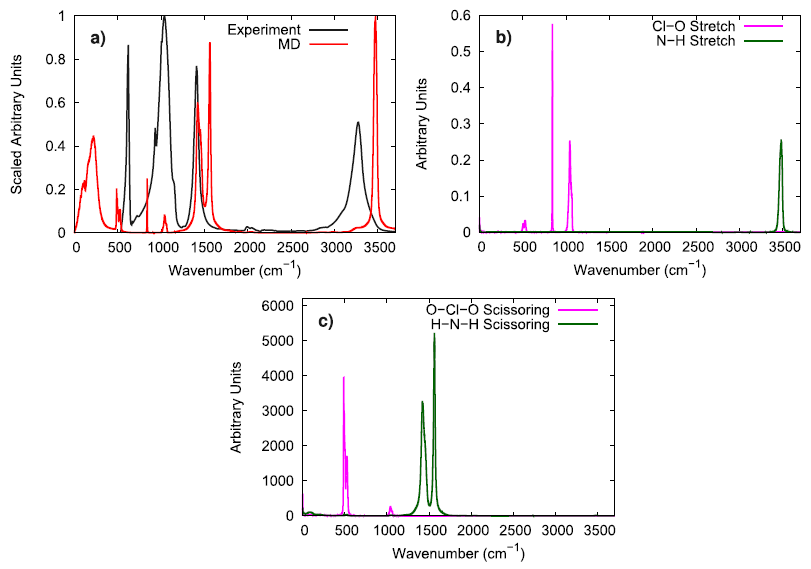
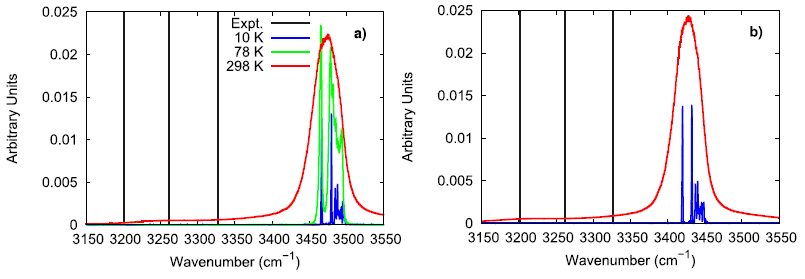
To further investigate the vibrational performance of the AP model, the temperature and time step dependence of the N–H stretching mode were assessed. At low temperature, the simulated AP shows three different N–H stretching modes which is expected for the three different hydrogen bonding environments that are experimentally reported. In comparison to Raman spectroscopy data, the simulated N–H stretching mode is significantly blue-shifted and the difference between hydrogen bonding environments is much smaller. The overly similar, simulated N–H stretching modes indicate that the N–H bonds in the force field model are not adequately susceptible to their local chemical environment to respond in a substantially different way, as seen in experiments. When lowering the time step from 1.0 fs to 0.5 fs, the N–H stretching mode is red-shifted, showing that a 1 fs time step is likely too large of a time step if concerned with the exactly vibrational details of the system. Still, the vibrational behavior in the simulated model is qualitatively consistent with experimental observations.
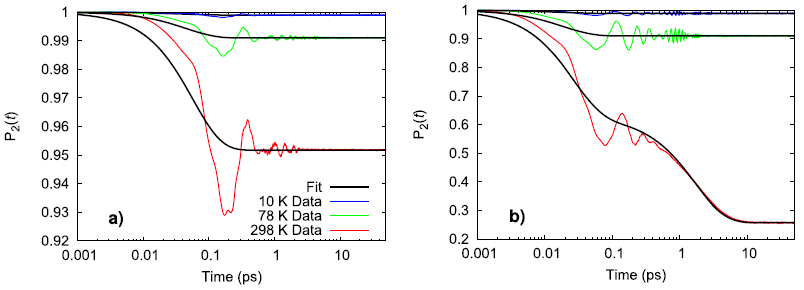
Experimentally, the perchlorate ion does not rotate between 10–298 K and only vibrates in place while the ammonium ion experiences the onset of nearly free rotation somewhere between 78–298 K. Using a second-order Legendre polynomial to describe the rotational behavior of the ions, the simulated ammonium perchlorate qualitatively agrees with the temperature trend observed in physical experiments.
Holistically, the modified AP model presented here is well-suited for reproducing trends in the material properties of AP between a temperature range of 10–298 K. The error in the quantitative reproduction of experimentally observed values by the model is generally ~5% or greater; however, this is not necessarily a significant issue as classical molecular dynamics simulations are frequently more appropriate for revealing trends and explaining physical phenomena rather than accurately providing quantitative estimates.
Current and Future Work
The development and validation of the models presented in this work are critical first steps in establishing classical molecular models for simulating solid propellant. Currently, validation of an AP-HTPB force field is being performed to match experimentally observed interfacial tension values between AP and HTPB. Once complete, interfacial phenomena important to describing the debonding process of HTPB from AP will enabled. In the future, it is important to develop a model for HTPB that is coarse-grained enough that long time scale simulations are possible for assessing mechanical properties on time scales accessible by experiment so that the simulation results can be corroborated. Eventually, the classical models developed as a result of this work will have degradation products introduced to the polymeric binder to assess how the material properties change as a function of how much thermo-oxidative degradation has occurred.
Acknowledgments
Support for this work was provided by the Air Force Office of Scientific Research under Contract AFOSR FA9550-18-1-0321. This work was supported in part by high performance computer time and resources from the DOD High Performance Computing Modernization Program as well as the Center for Research Computing (CRC) at the University of Notre Dame. The Materials and Process Simulation (MAPS) code was used for generating initial HTPB configurations. The support of Scienomics is acknowledged (Scienomics SARL, Paris, 2021).