Do you like the excellent Atom source editor, but find it to be slow AF? The problem is that Atom has too many default enabled packages and honestly some of them are poorly written. Go into your preferences and disable autocomplete and languages you don’t need…
New paper.
Lots of great stuff to follow 🙂
http://pubs.rsc.org/en/content/articlelanding/2014/SC/C7SC04934J#!divAbstract
How to generate Neural Network Training samples.
A new pre-print about sampling methods from John Kun & the gang.
https://arxiv.org/abs/1712.07240
New Pre-Print
https://arxiv.org/abs/1711.06385
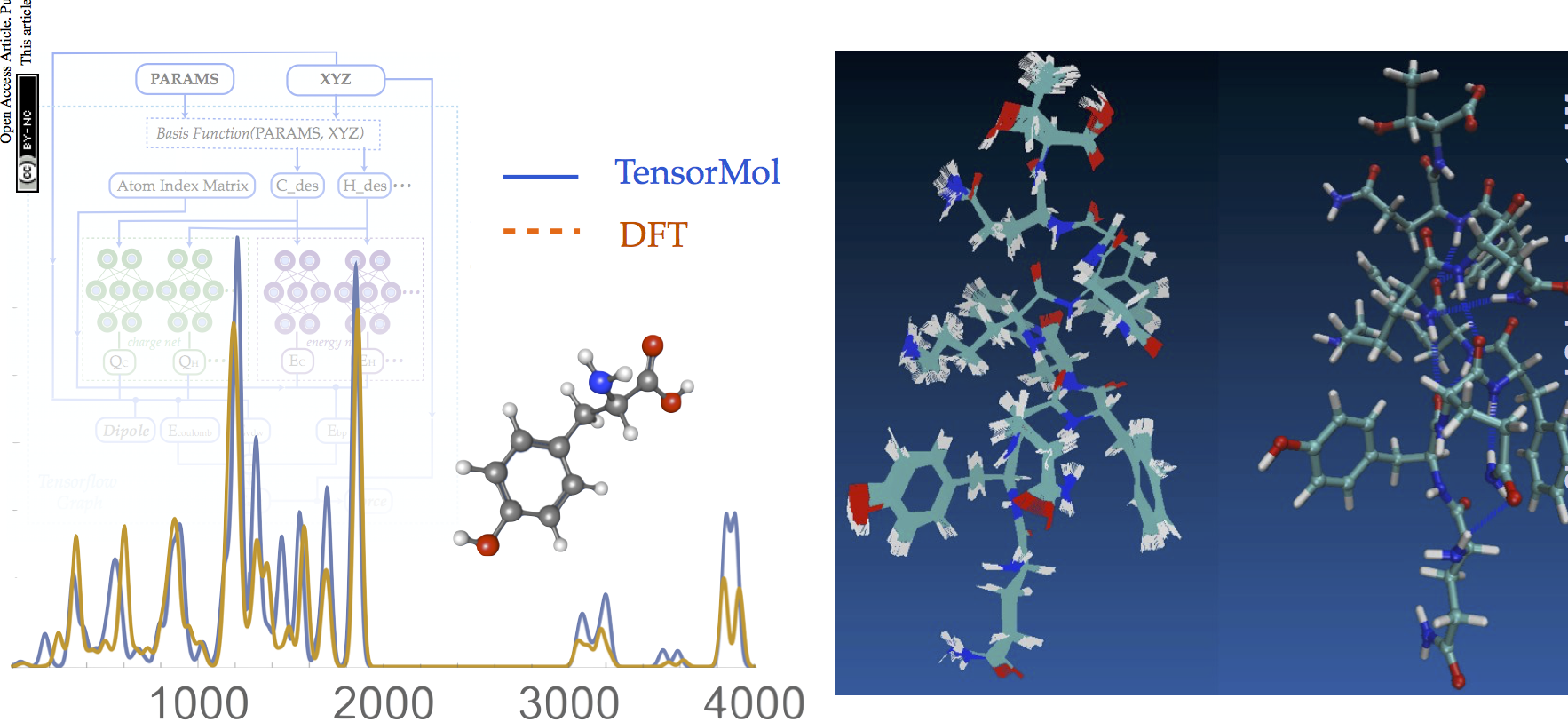
TensorMol0.1
TensorMol0.1 has been released, our package of Neural network model chemistries. It’s been a fun year putting it together. Give it a try.
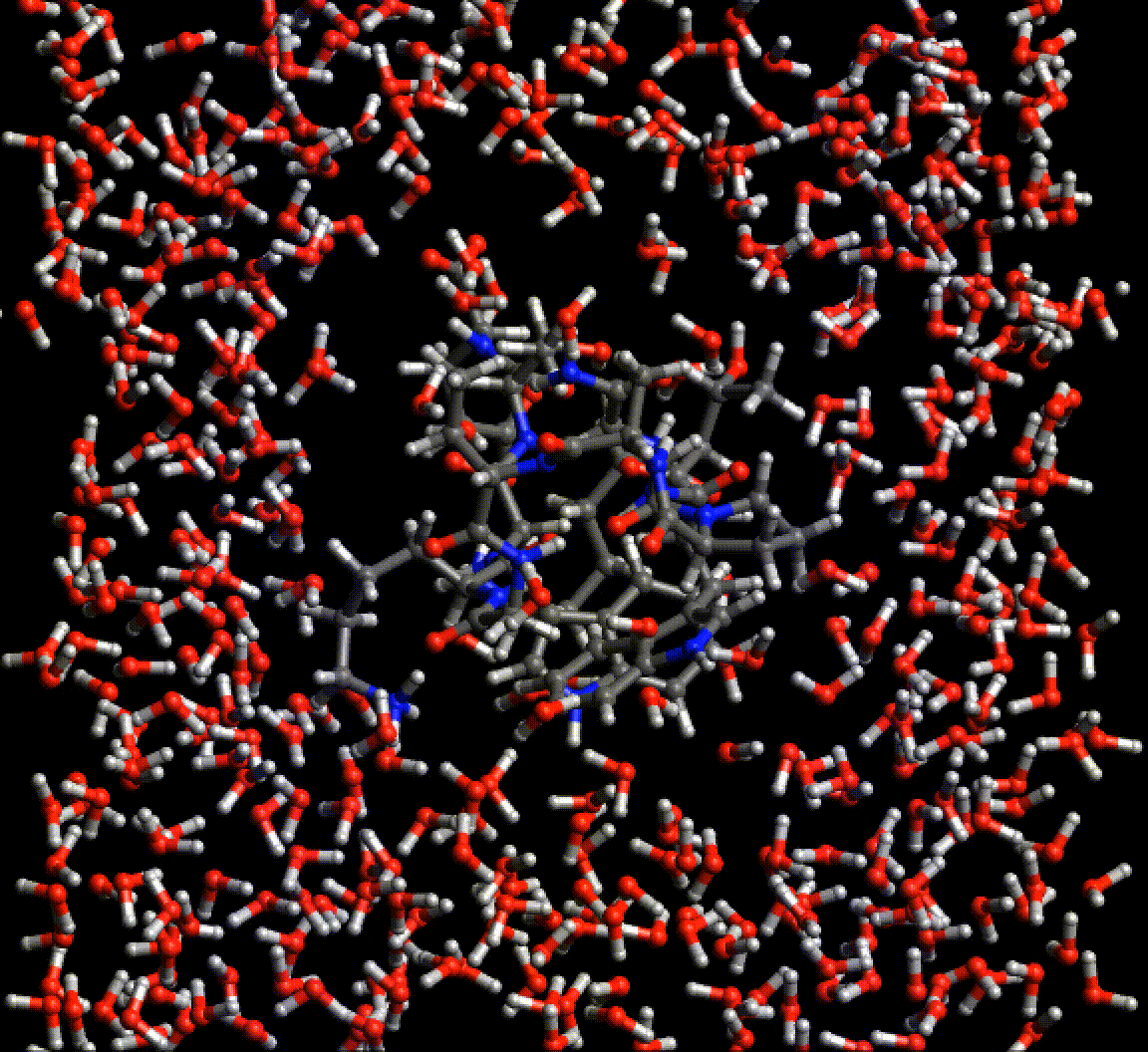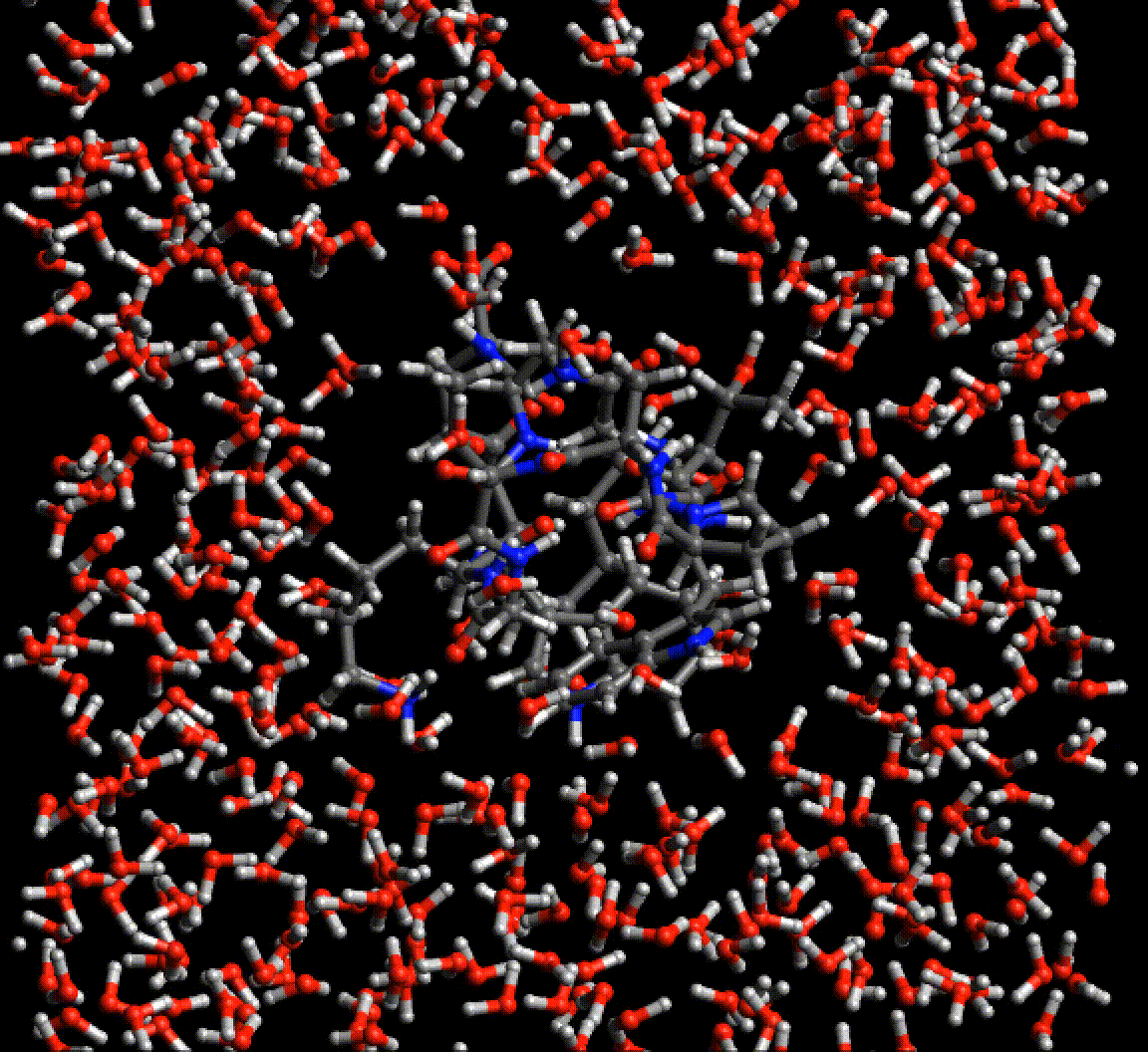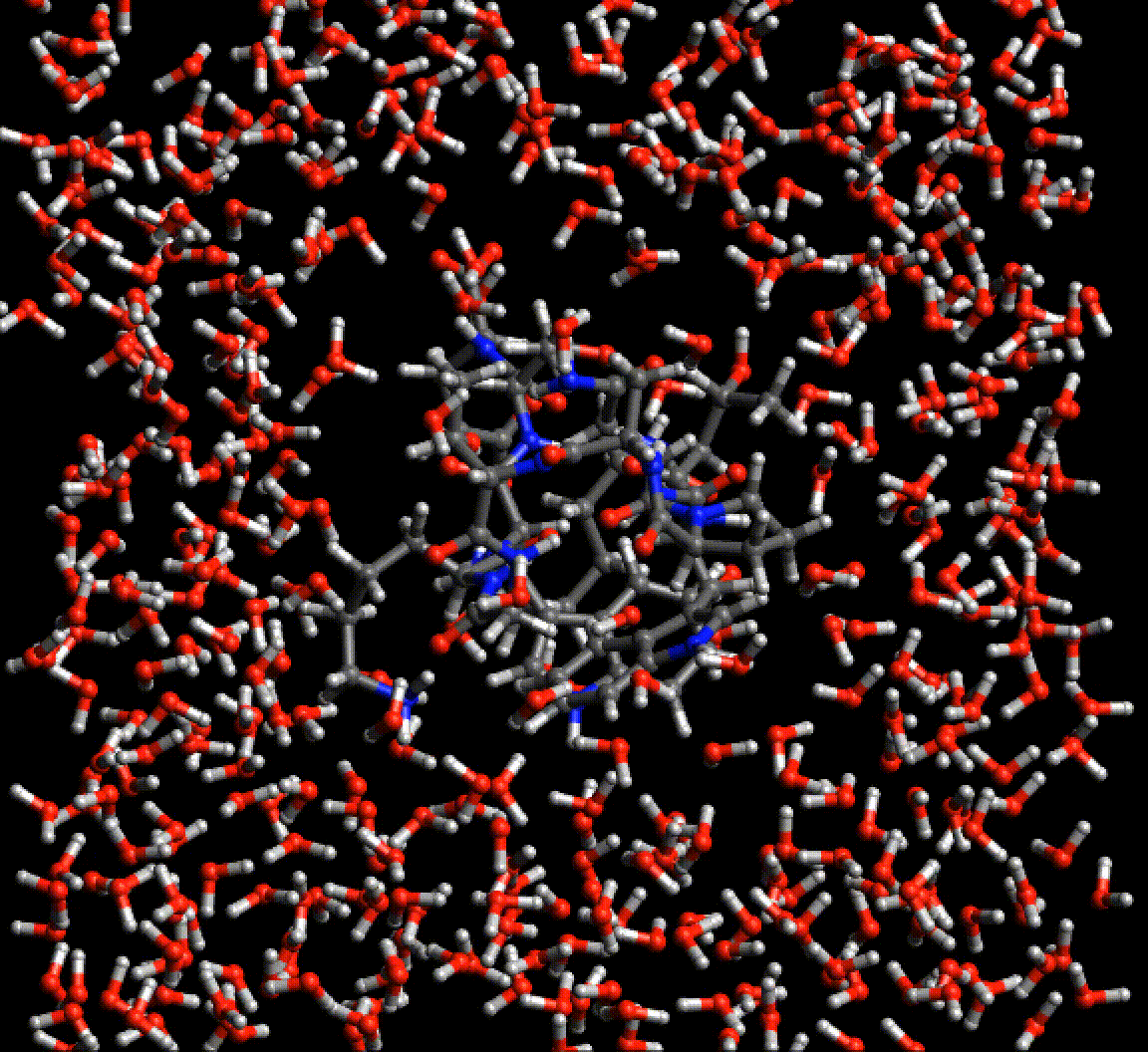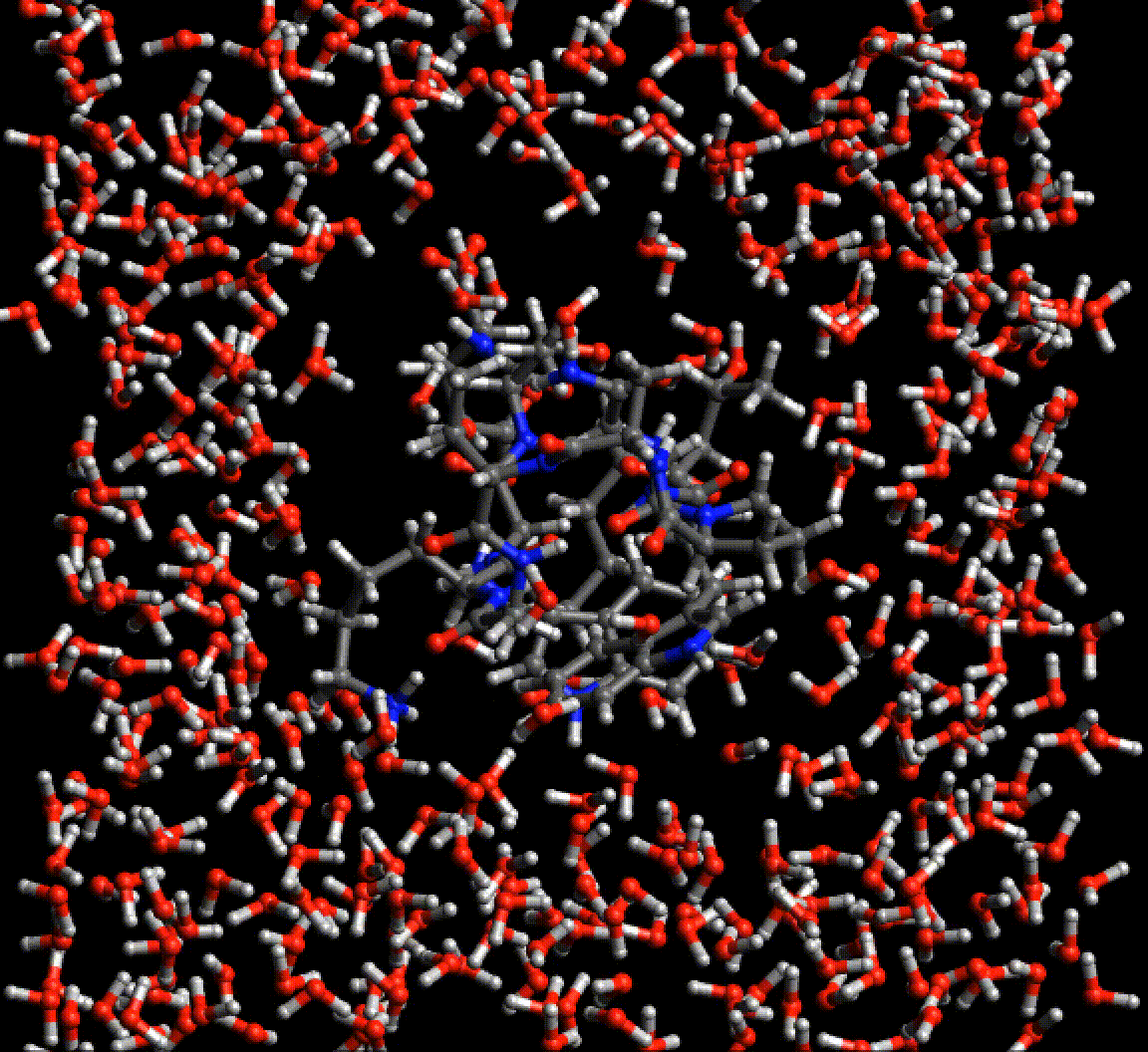
TensorFlow Tricks.
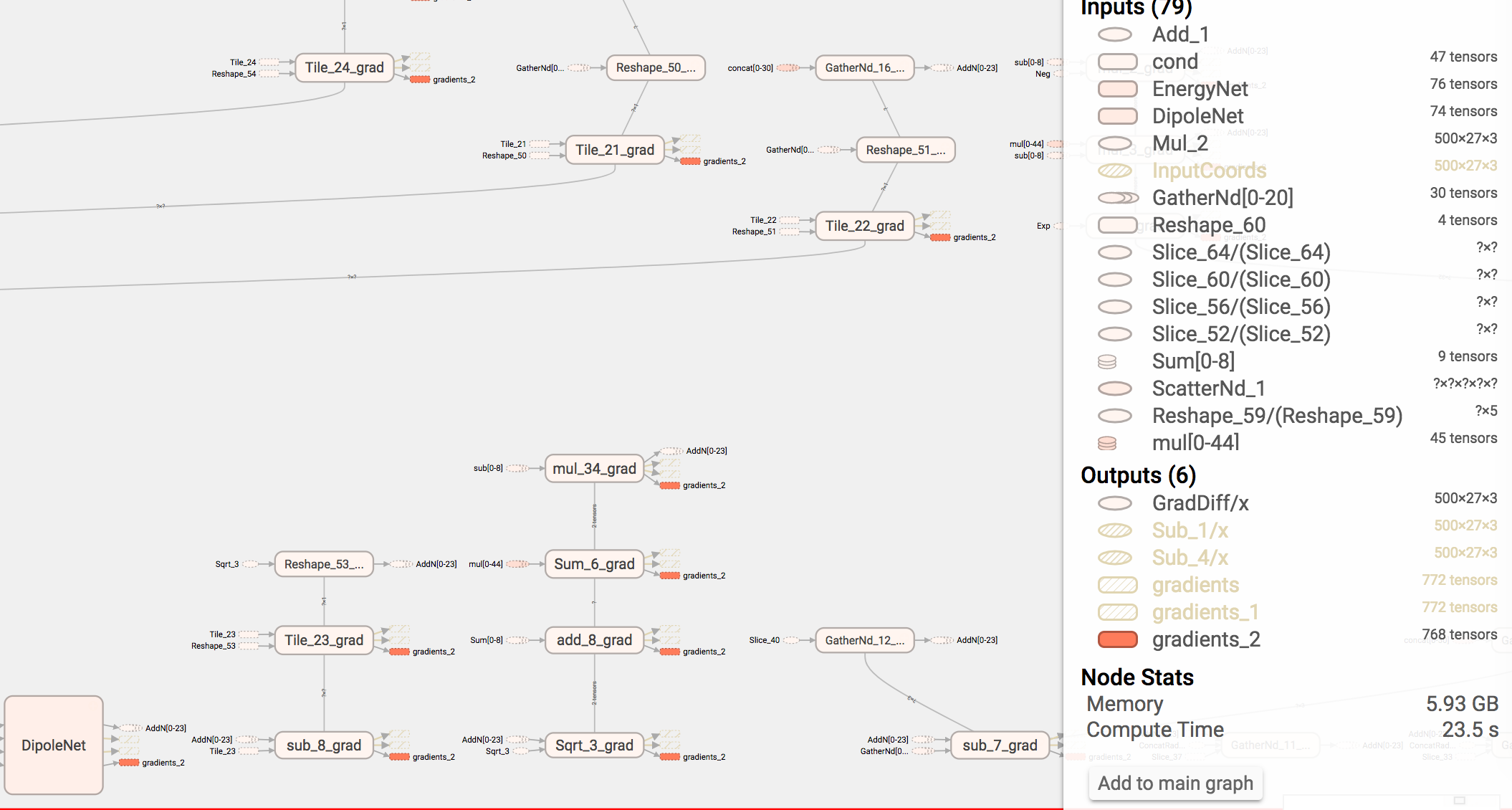
Just wanted to relay a little information which was useful. So we are producing a really complex neural network chemical model with hundereds of nodes and layers. This lead to only being able to (initially) fit small double-precision batches on the GPU. I wanted to explain how we debugged this….
Step 1: Add full profiling to the FileWriter and Graph Summaries.
self.options = tf.RunOptions(trace_level=tf.RunOptions.FULL_TRACE) self.run_metadata = tf.RunMetadata() self.summary_writer.add_run_metadata(self.run_metadata, "init", global_step=None)
Step 2: launch TensorBoard while profiling a training run.
tensorboard --logdir=PATHTONETWORK
Step 3: Point your browser to the locally hosted webpage tensorboard spins up. Pour over the graph and look at the memory usage of each fragment of the graph. naturally all the memory usage occurs mostly in the gradients which TensorFlow generates for training operations or the nuclear gradient for dynamics. In our case we learned that scatter_nd() which achieves the same effect as SparseTensor(), uses about two orders of magnitude more memory by default. Simply using SparseTensor instead fixed our issues. Happy hacking.
New Paper Accepted.
A collaboration with the excellent Kuno lab will appear in JACS:
“Origin of the Size-Dependent Stokes Shift in CsPbBr3 Perovskite Nanocrystals”
Author(s): Brennan, Michael; Herr, John; Nguyen-Beck, Triet; Zinna, Jessica; Draguta, Sergiu; Rouvimov, Sergei; Parkhill, John; Kuno, Masaru
New paper & Telluride.
Kevin and Triet’s paper applying Manby-Miller embedding to realtime TDDFT will appear in JCTC. Also if you’re in the neighborhood of Telluride, see you at the picnic in an hour or so 😛
-John
TensorMol on a Raspberry Pi3!
Popular Press for BIM-NN
https://www.chemistryworld.com/news/neural-network-predicts-bond-energies-like-a-pro/3007598.article
New Paper.
Bond Energies from a Bonds-in-Molecules Neural Network will appear in JPCL
http://pubs.acs.org/doi/abs/10.1021/acs.jpclett.7b01072
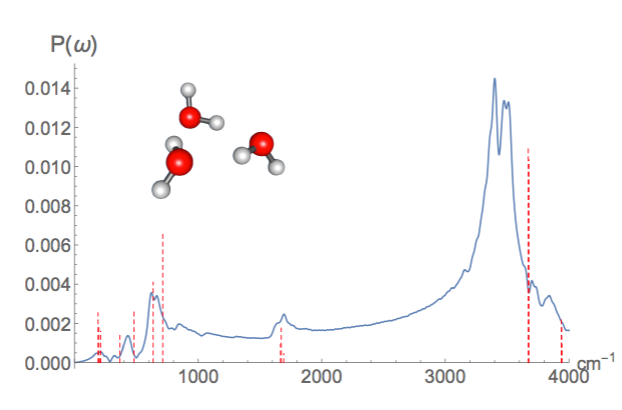
Now that IR spectra from Molecular dynamics is working, TensorMol is just about ready for a 0.1 Release! Below is a IR spectrum of water trimer made from realtime dynamics in TensorMol, compared with RI-MP2 stick spectrum.
The Lennard Jones Fluid with TensorMol
First in a series of videos about TensorMol. Rudimentary molecular dynamics of the LJ fluid. Automatic Differentiation forever!
https://www.youtube.com/watch?v=Qn2lYP9aadg&feature=youtu.be
Talk at MSU tomorrow.
I’ll be giving a seminar at MSU tomorrow. Congrats to Ben Levine on the JPC lectureship!
(PS- thanks Ben, Piotr and all of MSU’s fantastic Chem department for a very stimulating visit!)
New Pre-Print
Fun lil paper. One of the most frustrating things about being a theoretical chemist is when someone asks you to calculate something which is not physically well-defined (such as a bond energy). Neural networks can help us solve these problems, by learning the same thing the person is asking for, a number to match a heuristic intuitive concept. ArXiV for details:
Look at the K-Supercomputer…
The K Supercomputer has been operating since 2011, drawing 12.6 MW of power for ~88 thousand cores. It sits at the end of a rail-line with it’s own stop, in a shrine-like building next to a zoo. May not be the newest thing, but it still kinda makes a scientist wanna go ‘derp’.

APS, Kobe, ACS etc!
Just returned from New Orleans where our work was presented at the APS, next week headed out to Kobe, for the RIKEN AICS workshop talking about machine learning and then it’s back to the bay for the ACS meeting.
Congratulations Triet.
Triet was awarded an Eiler’s Graduate fellowship to support her work on Perovskites. Congratulations Triet.
New Paper accepted.
| The Many-Body Expansion Combined with Neural Networks |
K. Yao J. Herr and J. Parkhill, JCP 2016 10.1063/1.4973380
Upcoming Talks.
We have done a little cleanup of our Github and transitioned a bunch of private repositories to public repositories, and moved code from bitbucket onto github. I’ll be traveling a bit in the winter and early spring. Look for me at:
IPAM – MPSWS4 (San Diego December 5-8, 2016)
ACS San Francisco (April 2017)
Research Image of the Day

New Preprint.
http://arxiv.org/abs/1609.07072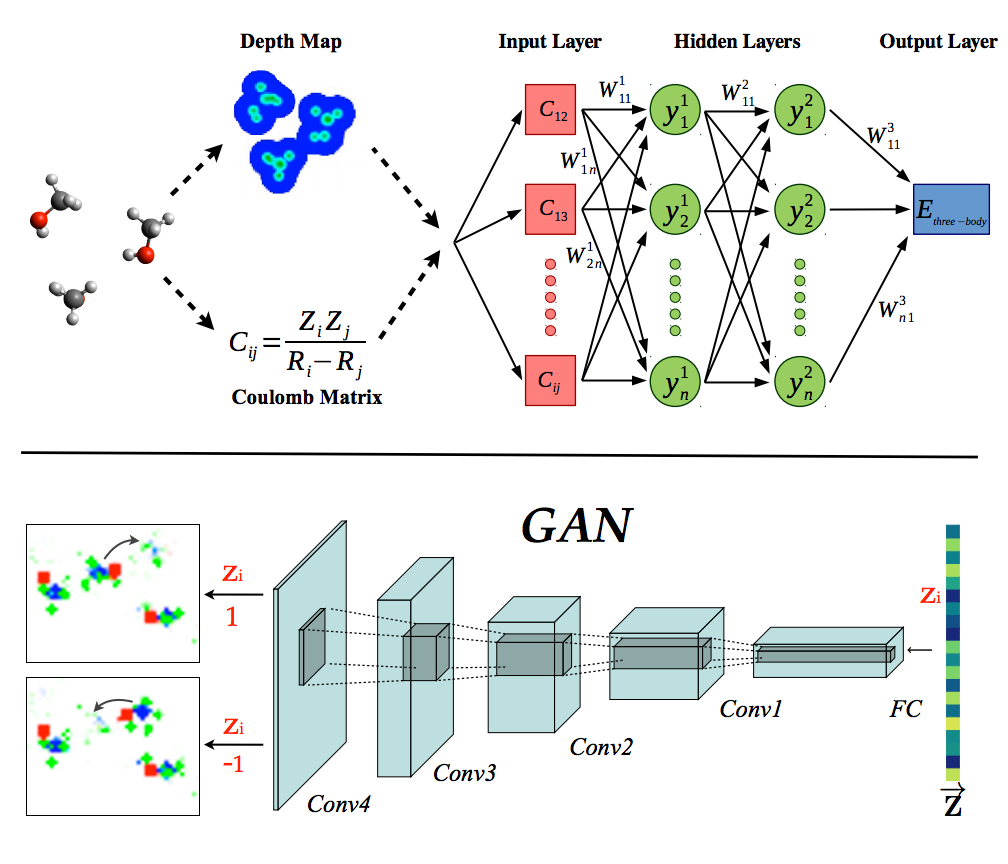
Gorgeous New data.
To be explained at a later date! Hint it’s C4H4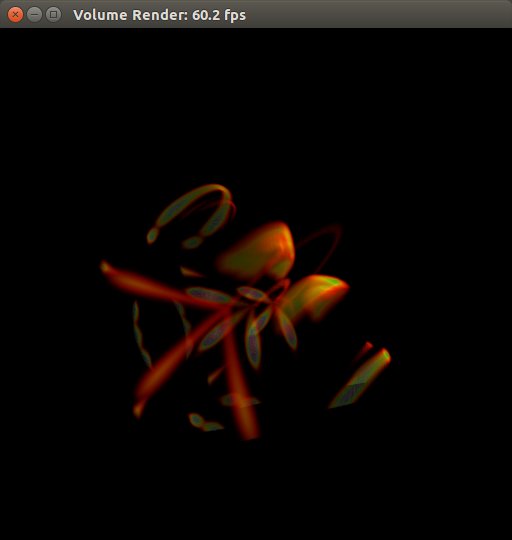
CTTCVII
Krakow is lovely! Talk tomorrow at 9am. If you’re in the neighborhood drop by Jagiellonian University, which is the source of Marie Curie. 
Tacc2016

Speaking at tacc around noon. Krakow next week. Say hi!
New paper
ASAP in Journal of Physical Chemistry A:
Nonradiative Relaxation in Real-time Electronic Dynamics OSCF2: Organolead Triiodide Perovskite. Authors: Triet Nguyen-Beck, John Parkhill.
Heidelberg@ND
This 4th of July week we’re hosting the Heidelberg@ND computational summer school. Thanks everyone for participating.

First LiveSlides presentation
Narrated by John Parkhill. Watch it here!
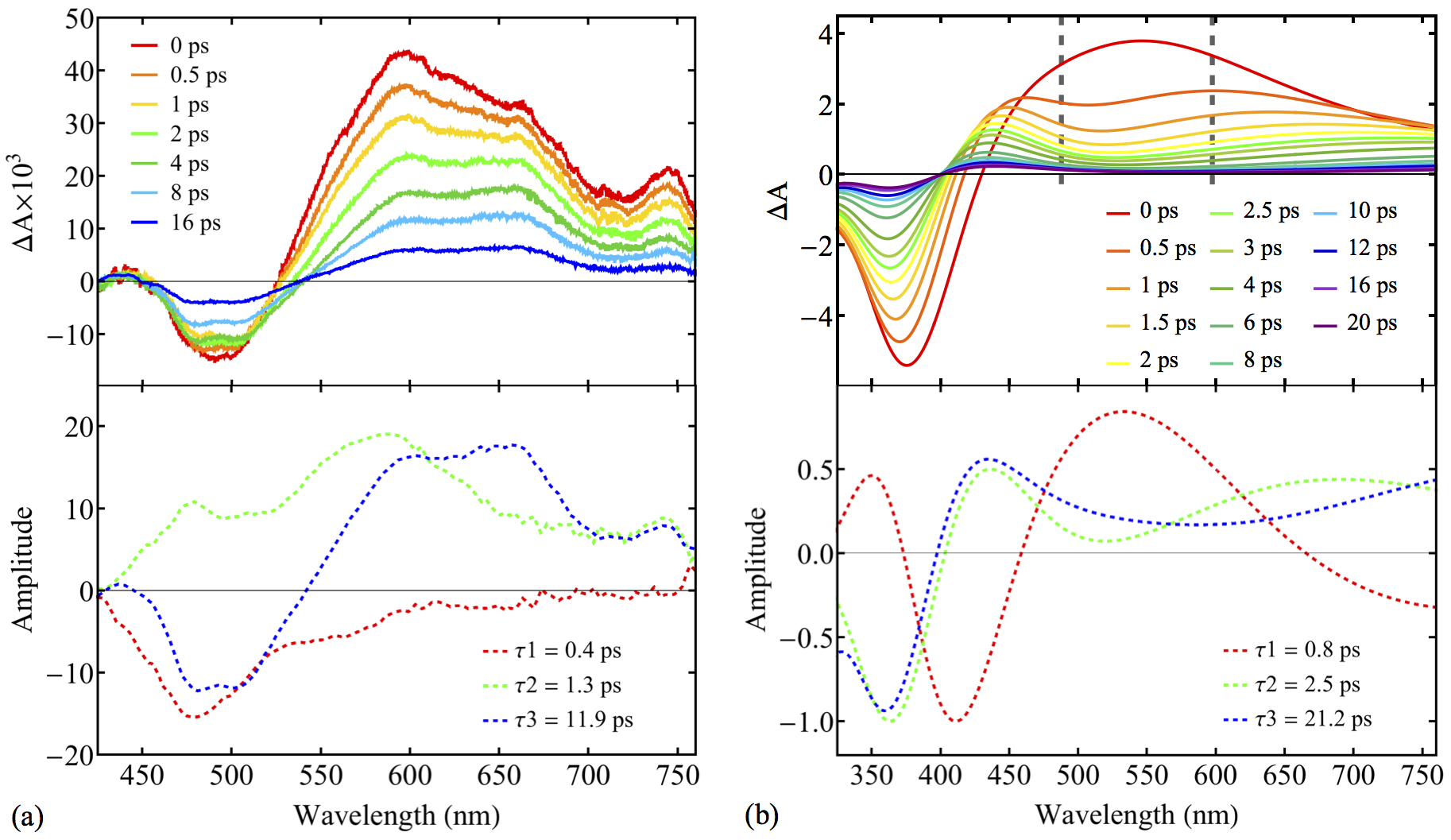
New paper.
“Black-Box, Real-Time Simulations of Transient Absorption Spectroscopy” will appear in JPCL. Authors: Triet S. Nguyen, Joong Hoon Koh, Susan Lefelhocz, John Parkhill.
ACS
Triet’s poster for the ACS in San Diego on Transient Absorption spectroscopy is looking great. It’s in the Sci-Mix section so take a look if you’re in the neighborhood. I’ll be speaking in three program items (all condensed into one day). Here’s what’s happening:

Heidelberg@ND
Image
A page announcing and collecting information for the Heidelberg@ND summer school is posted. This is a summer school opportunity for students interested in applying computational chemistry to experimental problems.

New Publication.
Kun’s Convolutional Neural Network functional will appear in JCTC (10.1021/acs.jctc.5b01011).
Susi Lehtola Visit and Welcome Kevin & John
This week we were visited by Dr. Susi Lehtola from Martin Head-Gordon’s group at Berkeley. Susi’s doing a high performance implementation of PQ, PH etc. and seeing phenomenal speedups (>500x) over the old code. It looks like he’ll be only limited by orbital transformation time, and thus able to study huge systems. Of course the tradeoff is that vs. my original machine generated code (which was still large. ~200Kb files), Susi’s source files are so big they are making GCC squeal (who doesn’t wanna hear GCC squeal). John Herr and Kevin Koh have officially joined up as of a few hours ago welcome guys. 
It’s working…
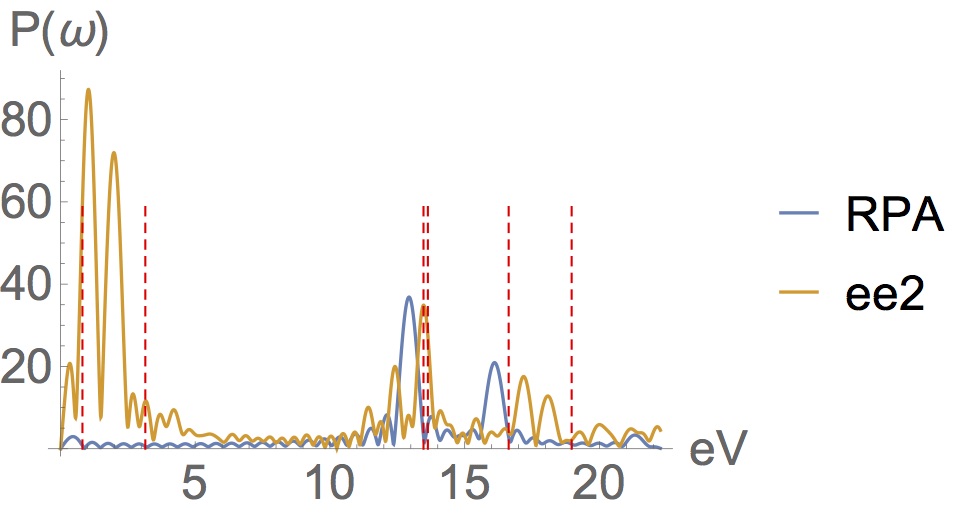
The RPA/TDDFT has many shortcomings, the most elementary being it’s inability to generate double excitations. Peaks are missing from the RPA spectrum and appear at the wrong energies. So here’s a difficult example where I’ve constructed a molecule which is strongly correlated (it’s bandgap is <1eV) and propagated it with our new EOM (ee2) & RPA. The exact transitions for this molecule occur at the red sticks. Despite the fact that it is based on the 1-RDM alone, is cheaper than Mp2, and despite the fact that it has no memory kernel, ee2 captures the missing states and double excitations. It provides a great improvement over RPA…
Halloween Stuff!
So much fun stuff has been going on that I’ve been neglecting to post. Let me summarize some developments:
- Figures from Triet’s research will be featured in the Shamrock Series 5k.
- Dr. Susi Lehtola will be visiting our group in November from Martin Head Gordon’s group in Berkeley.
- I’ll be charing a session about dissipative states at ACS San Diego, and participating in a Forum in the Comp Section.
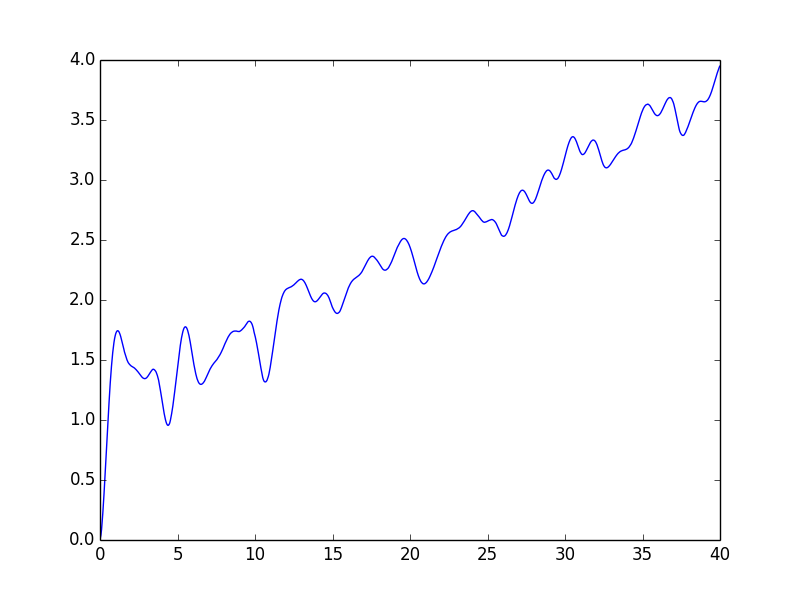
- Finally my favorite: I have realized a correlated electronic dynamics which preserves positivity and yet changes natural orbital occupations! This is a the culmination of years of effort and thought. This methodology can be used to study dynamics in fractionally occupied, strongly correlated systems (a hard problem). The method can be considered an improvement to RPA, and like it’s parent it is non-linear, actually fifth order in the 1RDM. The non-linearity is what allows this 1RDM method to preserve positivity while avoiding any memory kernel. The Kernel is effectively time dependent, but that time dependence is just a time-local function of the instantaneous density matrix. People who are interested in non-adiabatic TDDFT functionals: take note! performance of this EOM for excitation energies will be published shortly. Here’s a teaser plot of the electronic entropy growing with time (fs) . This is a strongly correlated 4 electron system where orbital occupations in the exact state should be roughly ~(0,0,0,0,1,1,1,1)…
New Pre-Print!
Anyone can now read about Kun’s convolutional neural network approach to kinetic energy functionals.
http://arxiv.org/abs/1509.00062
Welcome Rotation Students.
John Herr and Kevin Koh will be rotating with the lab for a few weeks, learning python and generally getting started. Welcome John & Kevin.
New Paper Accepted.
Triet’s second paper: “Non-adiabatic Dynamics for Electrons at Second-Order: Real-time TDDFT and OSCF2” will appear in JCTC. DOI: 10.1021/acs.jctc.5b00262
Postdoctoral Position Available.
Position requires proficiency in one of the standard computer programming languages used in science: C, C++, Fortran or Python, and a completed thesis on a topic relevant to quantum chemistry.
Student will have their choice of a broad range of topics in functional development, electronic or molecular dynamics, and correlation theory.
Interested parties should send their CV’s and best paper to john.parkhill@gmail.com.
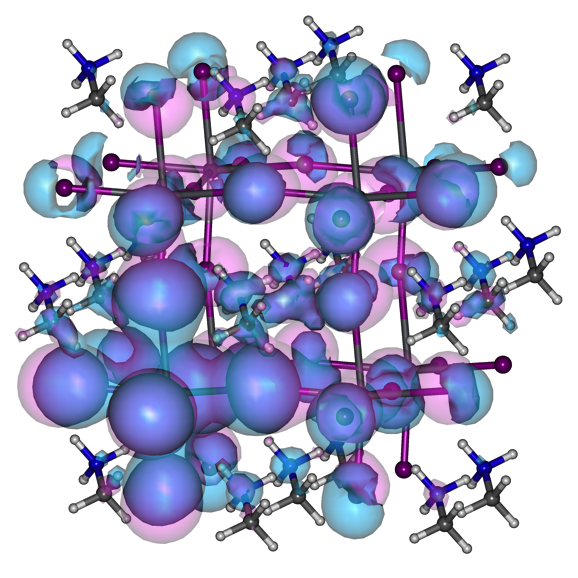
Put on your 3D glasses!
Just kidding! This is my failed attempt to visualize electron density (blue) and hole density (pink) of a CH3NH3PbI3 perovskite octamer, 200 fs after a laser impulse.
Fun Math Problem.
I just finished a really fun elementary math problem that I needed for my work; thought I’d share the answer. The problem is this: suppose you have 8 positive numbers (p,q,r,s,t,u,v,w), all co-prime besides possibly being equal. How many different types of equality ensure:
p+q+r+s-t-u-v-w = 0 (1)
For example one solution is (p=t, q=u, r=v, s=w). The answer is related to a “rotating-wave” or “secular” approximation for a many-particle density matrix in quantum dynamics. We can use sets of numbers which obey this condition to dramatically accelerate our many-body quantum dynamics calculations and treat correlation energy.
It turns out that the following expression of Kronecker deltas is 1 for every p,q,r,s… that satisfies (1) and zero otherwise, as you can easily verify. I’ll leave the derivation as an exercise to the reader :).

Paper up.
How electronic dynamics with Pauli exclusion produces Fermi-Dirac statistics.
Nguyen, Triet S.; Nanguneri, Ravindra; and Parkhill, John. The Journal of Chemical Physics, 142, 134113 (2015). DOI: http://dx.doi.org/10.1063/1.4916822
Scientific Art.
The best part about this blog is we can post attractive but unconventional scientific stuff. This is a lovely “table-of-contents” style graphic that Triet has made about our recently developed realtime dynamics code. It nicely sums up our efforts in this area. The data is real too.
Lead Halide Perovskite (PbI3-CH3NH3) Under Impulsive Excitation
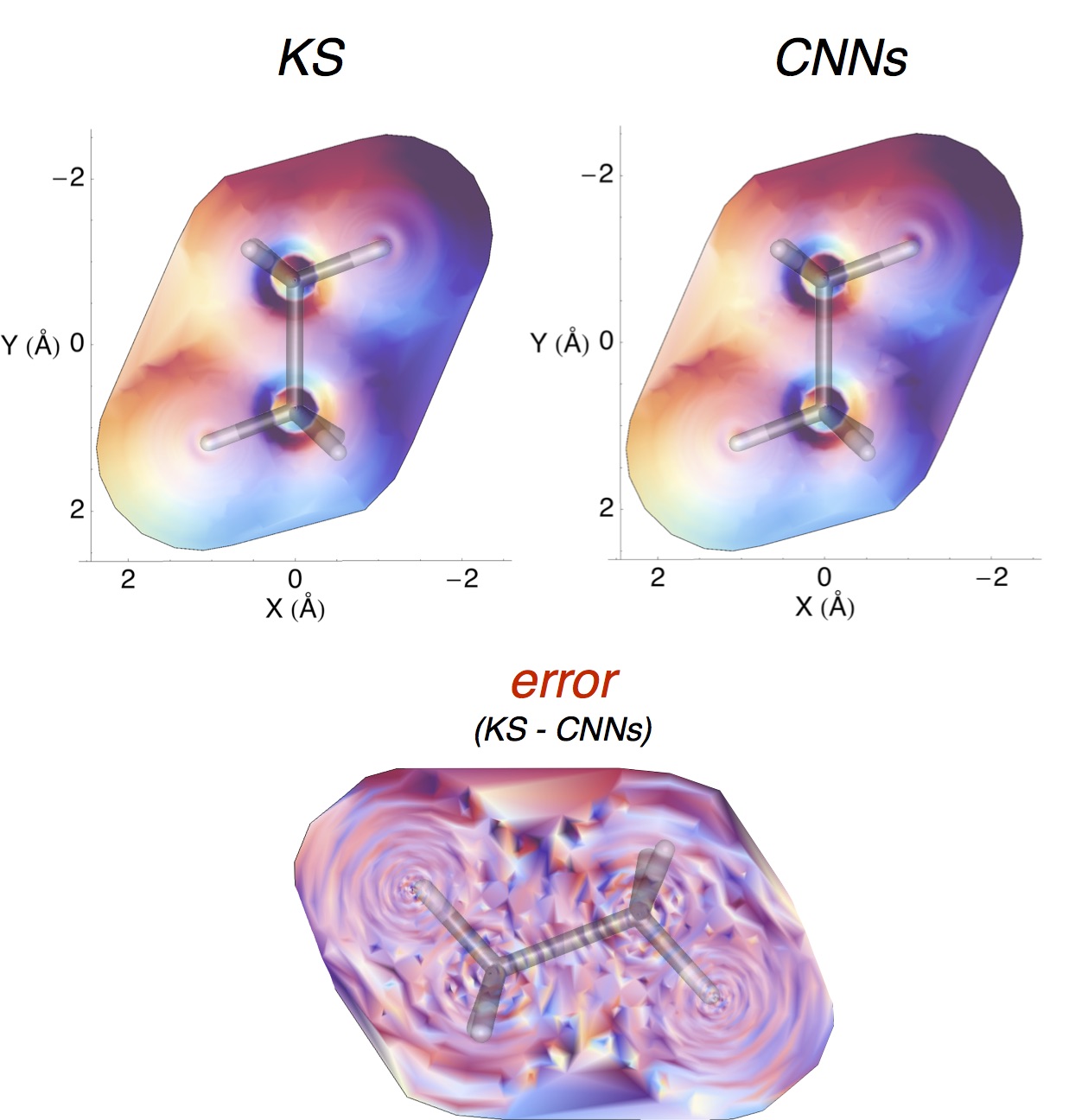
A broadband ‘light’ impulse of duration 0.07 atomic units is applied along the x-axis at time t=0. The electronic dynamics includes our dissipation theory, although this short time (~20fs) is not enough to watch the electron relax back to it’s ground state position. If you pay close attention to the atoms on the central line you can see the average ‘slosh’ of the density (on a timescale of about 1/frame which is roughly 1fs). The excitation is mostly due to rearrangement of the density onto lead from iodine. If you look at the surronding iodine atoms you can see their p-density rotate, responding to the central density oscillation, that’s what you’d call ‘dynamic screening’. (Click the image to animate)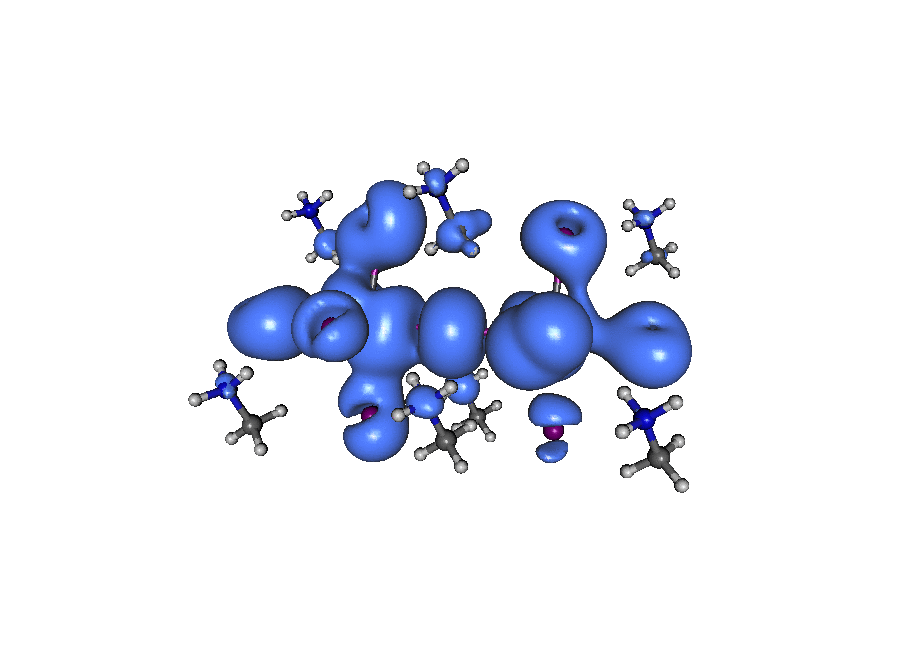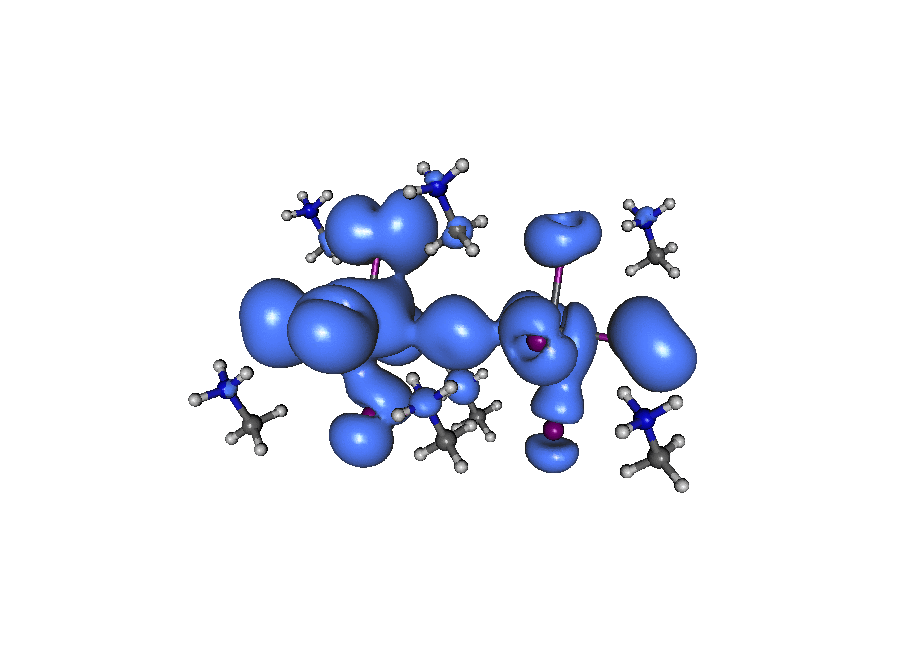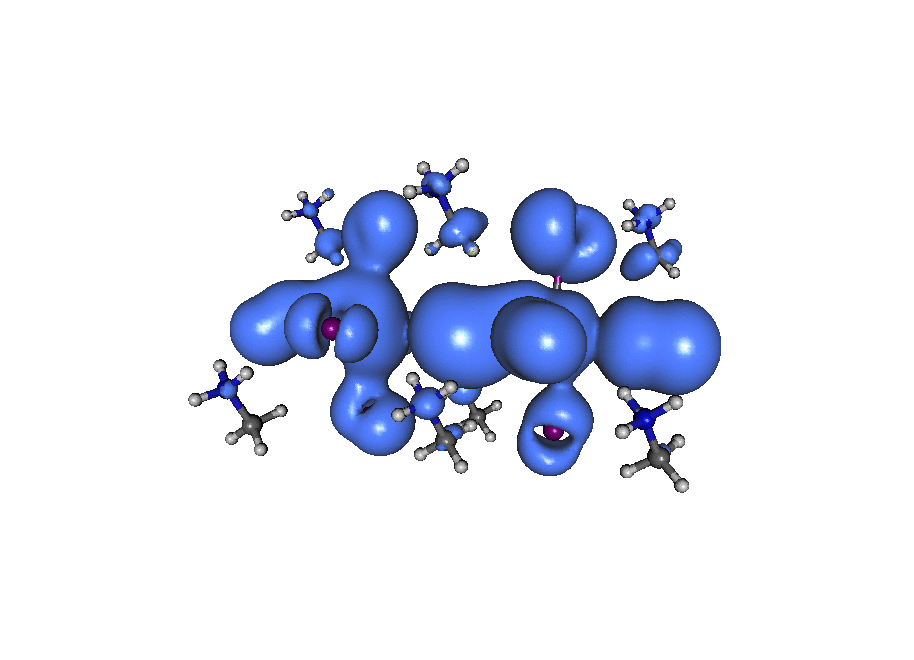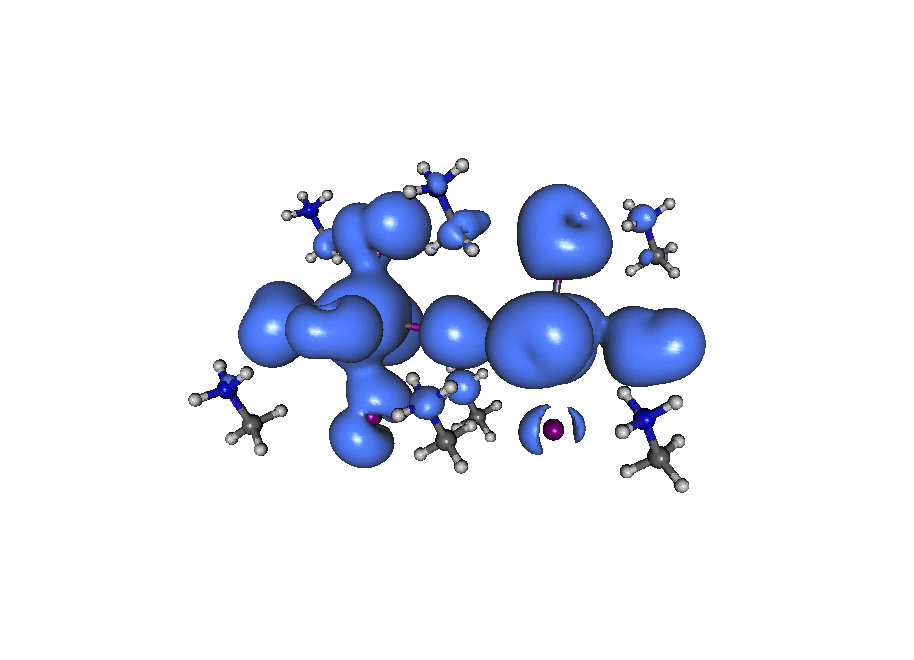
University Remembers Hesburgh
It’s unlikely that our research group would exist without the work of Father Theodore Hesburgh, a remarkably successful and dynamic personality who devoted his life to other people. We pay our respects (http://hesburgh.nd.edu/).
Benzene under resonant excitation.
Interesting how the sigma framework fills in first.
(click to animate if it’s not already moving.)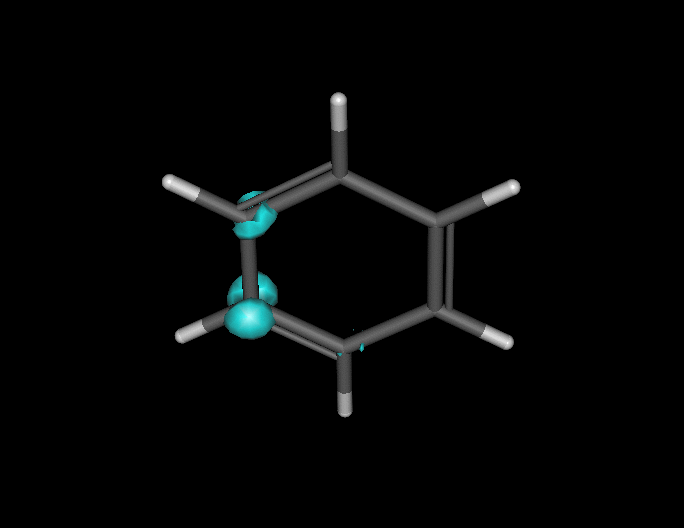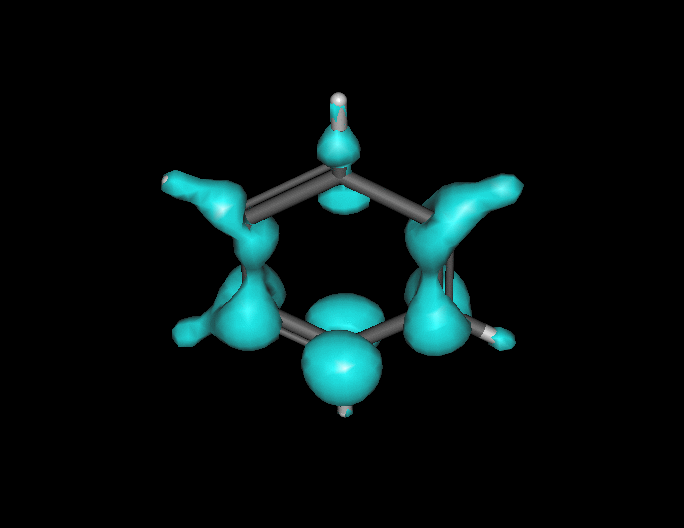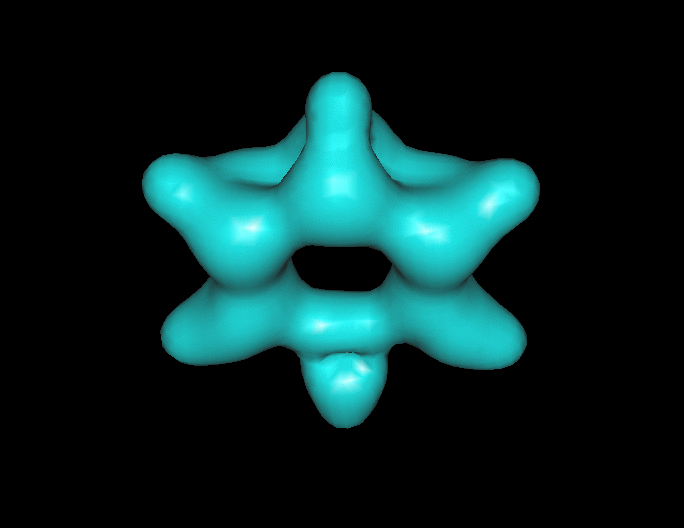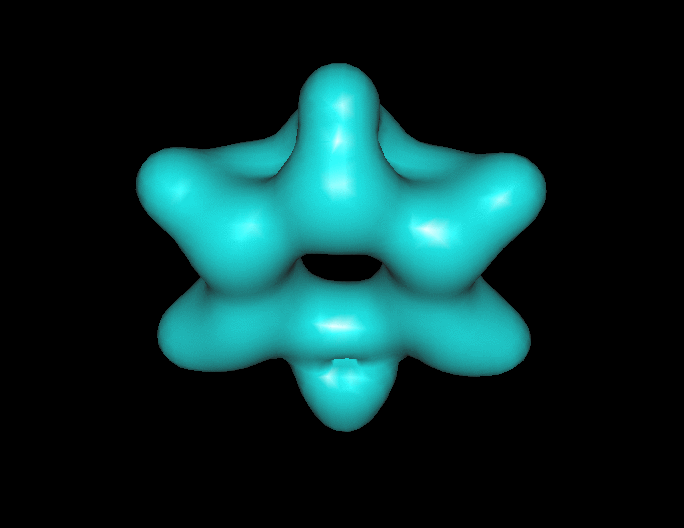 )
)
Upcoming Meetings.
Want to hear about what we’re up to? We’re going to cover a pretty large swath of territory over the next few months. If you’re in the neighborhood we love to meet old and new friends.
Feb 19th: Notre Dame Physics Seminar
March 22nd – 26th: ACS National Meeting in Denver (Symposium for Highly Polarizable Systems)
May 26nd – 29th: Many Body Interactions Workshop
July 5th -12: The Joint Heidelberg-Notre Dame Summer School in Computational Chemistry
July 13th-17th: Telluride Summer Schools
(also staying for Nonequilibrium Phenomena, Nonadiabatic Dynamics and Spectroscopy”, July 20–24, 2015)
Relaxation movies with Jmol.
Probably the easiest but not the most attractive way to generate an animated gif of electron density during a relaxation process is the combination of Jmol with imagemagik. You run our code to generate a molden file with densities in sequential orbitals, and then run the following Jmol script after opening that molden file and dragging this into the script console. I have shamelessly adapted this script from Google:
print “Dumping densities... ”
for (var i = 1; i <= 1000; i = i+4)
print i;
var filename = "movie"+("00000"+i)[-4][0]+".jpg”;
mo @i;
mo cutoff 0.000005
mo translucent 0.3;
mo Fill noMesh noDots frontOnly frontlit;
write IMAGE 600 400 JPG @filename;
frank on;
select *;
set fontScaling false;
end for
At that point in your Jmol has dumped .jpg’s into it’s directory which you can then convert into an animated gif by simply running the following command in that directory provided you have installed imagemagik. The animated .gif made below is the excess electron density (above the DFT ground state) caused by light irradiation along the x-axis of a hydrogen ring (click to see the animation in Chrome). “Energy” in the text of the plot is actually the time of the frame in atomic units. The occupancy of the excess electron is small because the applied field was ~0.0001 atomic units, and it oscillates while the excited states persists before it eventually decays to zero.
convert -delay 10 -loop 1 *.jpg animated.gif

Happy holidays.

Welcome new member Susan Lefelhocz
Susan is a Freshman at Notre Dame, and looking into some QM-MM issues. We also recently finished a computer algebra for the Mukherjee-Kutzelnigg Normal Ordering, and it’s Kun’s birthday. Happy Birthday Kun.
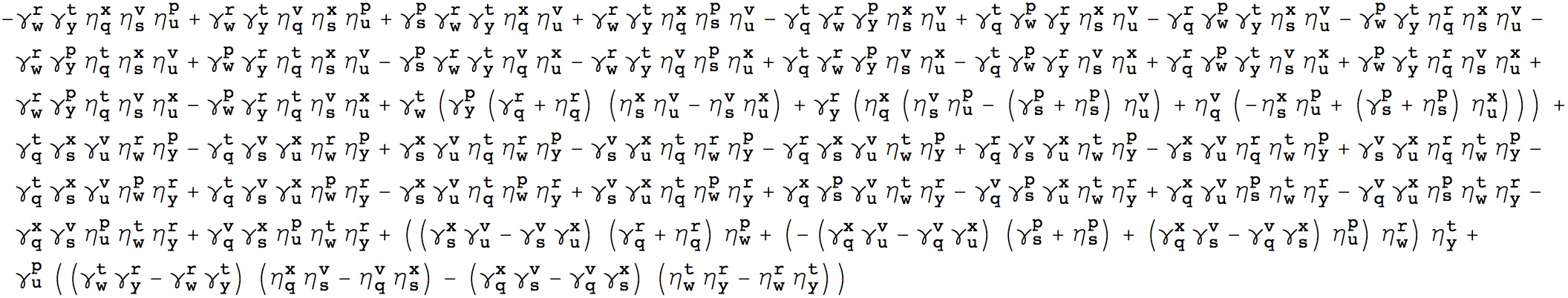
To equilibrate properly electronic relaxation rates depend on the density matrix!
In high dimensional systems very few exact results are known about quantum dynamics. One of the most important exact conditions we can try to satisfy is detailed balance, ie: dynamics should equilibrate to the correct statistical distribution, a Fermi distribution for electrons, at long times.
Lots of people are familiar with Surface Hopping, Redfield theory, and Ehrenfest dynamics, but actually you can’t use any of these methods to produce a Fermi distribution exactly. Based on our work simulating relaxation, we’ve actually been able to derive an equation of motion which does obey Fermi-Dirac statistics. In obtaining the derivation, we learned useful tricks that are going to help us treat mixed-states on the same footing as pure states. We’re super jazzed about these things.
There are cool experimental consequences, for example that non-radiative relaxation rates are not constant with time. You can check out the whole story on ArXiv for the time being:
